GC、GC-MS法定性定量法检测血液和尿液中阿片类毒品及其代谢物的含量
2016-07-02努尔艾力塔依尔毛新民车文森艾斯凯尔艾尔肯艾克拜尔热合曼
努尔艾力·塔依尔,毛新民,车文森,艾斯凯尔·艾尔肯,艾克拜尔·热合曼
(新疆医科大学1基础医学院法医教研室; 2中医学院,乌鲁木齐 830011; 3乌鲁木齐市公安局刑科所,乌鲁木齐 830000)
GC、GC-MS法定性定量法检测血液和尿液中阿片类毒品及其代谢物的含量
努尔艾力·塔依尔1,毛新民2,车文森3,艾斯凯尔·艾尔肯1,艾克拜尔·热合曼1
(新疆医科大学1基础医学院法医教研室;2中医学院,乌鲁木齐830011;3乌鲁木齐市公安局刑科所,乌鲁木齐830000)
摘要:目的建立检测血液、尿液中阿片类毒品及其代谢物含量的气相色谱(GC)法或气-质联用(GC-MS)法。方法尿样或血样中的阿片类毒品,加0.1 mol/mL (pH=6)磷酸缓冲溶液调节pH为5~6,将样品添加于固相萃取柱(SPE),用去离子水和0.1 mol/mL HCl淋洗,再次用 CH3OH洗涤SPE柱子,由含2%氨水的CH2Cl2/CH3CH2OHCH3混合液萃取提取,吹干,用BSTFA+TMCS(99∶1)试剂衍生化,以SKF-525a作为内标,利用色谱柱为DB-5MS的GC、GC-MS仪选择离子(m/z)扫描模式(SIM)和全扫描(Full Scan)模式进行测试分析。结果海洛因、吗啡、可待因、6-乙酰吗啡、6-乙酰可待因、乙基吗啡在25~400 ng/mL范围内与峰面积呈良好的线性关系,相关系数r>0.990 7,平均加内标回收率为95.9%~101.6%,相对标准误差(RSD)为2.13%~8.59% ,最低检测限(LOD)为1 ng/mL,最低定量限(LOQ)为25 ng/mL。案件样品尿液检测出可待因、吗啡成分、6-乙酰吗啡成分,可待因含量>400 ng/mL,吗啡为384 ng/mL,6-乙酰吗啡为278 ng/mL,血液检测量极小,确证了吸食海洛因的违法事实。结论本方法简单、快速、灵敏度高、准确定性定量,适用于吸毒或毒品滥用者血样、尿样的筛查与确认试验。
关键词:阿片类毒品; GC/MS; SPE萃取法; 衍生化; 海洛因
阿片类药物是指天然的或人工半合成的罂粟类生物碱以及其衍生物,含有二十多种不同的生物碱,其中吗啡、那可汀、罂粟碱、可待因、蒂巴因、那碎因和苄异喹啉类7种生物碱已被提取分离[1]。目前以海洛因为代表的阿片类毒品严重威胁人类健康。据联合国毒品与犯罪问题办公室(UNODC)并报告,戴着全球滥用违禁药品的吸毒人数已超过3.25 亿,占世界人口4.64%。20世纪80年代初全国登记在册的海洛因等阿片类滥用者约为7 万人;而截至2014年4月底已达到258 万人。在今后相当长的一段时间内,海洛因滥用仍是人类面临的最大的挑战[1]。虽然国际国内的毒品形势正在从传统的鸦片类毒品向兴奋类新型毒品转变,但目前我国毒品泛滥比较严重仍是海洛因[2]。生物样品(血液、尿液)中毒品成分及其代谢物的检测方法种类很多,目前最常用的是用胶体金初筛法(免疫法)和气-质联用(GC-MS)仪或LC-MS。三重四极杆质谱仪已经在部分研究和鉴定部门普及[3-4],但由于仪器昂贵,操作成本和人员要求高,在我国的普及率还不高,因此多采用气相色谱-质谱(GC-MS)方法。复杂的生物样品不能直接仪器检测,需要前处理的,通过固相萃取法对复杂生物样品进行分离、提取,进行衍生化,产生硅烷化产物,其极性降低,挥发性增强,沸点降低,可以得到满意的检测结果[5-7]。本研究通过固相萃取SPE法分离、提取生物样品中的阿片类毒品,用三甲基氯硅烷硅烷化试剂(BSTFA+TMC、99∶1)衍生化,采用气相色谱-联质谱(GC-MS)仪,选择离子(m/z)扫描模式(SIM)和全扫描(fullscan)模式对生物样品中阿片类毒品及代谢物进行定性定量分析。通过此方法对吸毒者生物样本进行定性定量分析,现报道如下。
1仪器和试药
1.1仪器气相色谱/质谱联用仪(气相色谱为安捷伦7890A,质谱为Waters Quattro micro),气相色谱仪(GC 2010 PLUS,日本岛津公司),低速大容量离心机(TDL-508型,上海安亭科学仪器厂),多功能振荡器(XH-J,金坛市医疗仪器厂),氮吹仪(HGC-12 A,天津恒高科技有限责任公司),电子分析天平(AB135-S型,瑞典METTLER TOLEDD公司),气相色谱色谱柱子(RESTEX-RTS5,美国RESTEX公司),气质联用仪色谱柱子,(DB-5MS,安捷伦公司),Bond Elut Plexa固相萃取小柱SPE(130 mg,3 mL,购自Varian公司)。
1.2试药阿片类毒品标准样品二乙酰吗啡盐酸盐(批号171206)、盐酸吗啡(批号171256)、磷酸可待因(批号171203)、SKF-525a(批号567300)均由公安部物证鉴定中心提供,海洛因(1.0 mg/mL,批号 1183002)、6-乙酰可待因(1.0 mg/mL,批号17209)、6-乙酰吗啡(1.0 mg/mL,批号 171207)、乙基吗啡(1.0 mg/mL,批号 920301)均购自于Sigma公司,BSTFA+TMCS(99∶1)衍生化试剂(Sigma公司,批号LB97094),甲醇、乙醇、丙酮、乙酸乙酯、异丙醇均为色谱纯,四氯酸、盐酸、氨水、二氯甲烷为分析纯。
2方法和结果
2.1标准物质储备液的配制分别准确称取海洛因、吗啡、可待因标准物质10 mg,用10 mL甲醇溶解配制成1.0 mg/mL的标准储备液。6-乙酰可待、6-乙酰吗啡、乙基吗啡均为1.0 mg/mL液体状安瓿瓶,精确配制成500、100、10、1 μg /mL的甲醇系列储备液,置于-20℃冷冻室中保存。0.1 mol/mL(pH=6)磷酸缓冲溶液的配制:称取6.82 g KH2PO4(MW=136.09 g/mol)于500 mL试剂瓶中,用450 mL纯水震荡溶解,并先用10 mol/mL KOH溶液加约3 mL,再滴加1.0 mol/mL KOH溶液,调节溶液pH 值为6.0(0.1),使用pH酸度计测定与确定缓冲溶液pH 值。若加KOH过量pH值偏高,则用1 mol/mL HCl溶液来调节,最后加水稀释至500 mL定容。含2%氨水的CH2Cl2/CH3CH2OHCH3混合液(80∶20) 的配制:80 mL二氯甲烷和20 mL异丙醇混合,除去200 μL混合液,再加200 μL氨水,摇匀混合即得。0.1 mol/mL HCl溶液的配制:量取4.135 mL的37% 、浓度为1.19 g/mL浓盐酸溶液,放入500 mL试剂瓶中加水稀释至刻度位置。0.8 mol/mL高氯酸(HClO4)的配制:量取3.5 mL的70%、浓度为1.66 g/mL的HClO4溶液,放入50 mL容器中加水稀释至刻度位置。
2.2色谱条件色谱柱为安捷伦DB-5MS(30 m×0.25 mm×0.25 μm),温度范围-60℃~325℃(最高值360℃)。载气:高纯氮气 ,流速1.5 mL/min;程序升温模式:柱温110℃(恒温),保持2 min,再以10~15℃/min速度升到280℃(程温),保持10 min;进样口温度:280℃;气化室温:270~300℃;检测器温: 290~300 ℃;质谱条件:EI电离源,电子能量70 eV,离子源温度250℃。调谐方式为自动调谐,倍增电压1 073 V,发射电流100 μA,扫描范围40~500 m/z。
2.3样品前处理
2.3.1血样前处理用移液枪吸取2 mL血液(已加内标的待测样或已加内标的空白血样)放入10 mL离心试管中,添加 20 mg 偏重亚硫酸钠(Na2S2O5)、2 mL 蒸馏水、2 mL 0.8 mol/mL高氯酸(HCLO4),混匀,3 000 r/min离心10 min,将上层有机相转移至另一已经标签好的10 mL试管中,再加入2 mL(pH=6)的磷酸缓冲溶液,用1 mol/mL的KOH溶液调节pH值为5~6。此溶液将进一步进行固相萃取(SPE)[注:在血样内标最终浓度应在100 μg/mL,在2 mL血样中添加200 μL 10 ng/μL内标物溶液(SKF-525a)]。
2.3.2尿样前处理用移液枪吸取2 mL尿样(已加内标的待测样或已加内标的空白尿样)放入10 mL离心试管中,加入5 mL(pH6)的磷酸缓冲溶液,混匀,3 000 r/min离心10 min,用1 mol/mL的KOH溶液或0.1 mol/mL或1 mol/mL H3PO4溶液调节pH值为5~6。该溶液将进一步进行固相萃取(SPE)[注:在尿样中内标最终浓度应在100 μg/mL,在2 mL尿样中添加200 μL 10 ng/μL内标物溶液(SKF-525a)]。
2.3.3样品固相萃取(SPE)法处理(1) 活化SPE柱子:分别用3 mL甲醇和3 mL 0.1 mol/mL(pH=6)的磷酸缓冲液活化,即先用3 mL甲醇溶剂添加SPE柱子中,加3 mL 0.1 mol/mL(pH=6)的磷酸缓冲液淋洗。(2) SPE柱子中填充样品:将上述前处理过的血样或尿样加入已活化好的SPE柱子中,以1 mL/min流速过滤样品溶液。(3) 洗涤SPE柱子:先用6 mL去离子水淋洗,然后用3 mL 0.1 mol/mL HCl溶液洗涤,减压抽干2 min,最后用9 mL CH3OH再次淋洗SPE柱子。(4)萃取提取:新配制的含2% 氨水的CH2C2/CH3CH2OHCH3混合液(80∶20)2 mL加入SPE柱中,在常压条件下,将柱子中的毒品成分,洗脱2 mL安瓿瓶中。(5) 吹干提取液:将2 mL安瓿瓶中的洗脱液在氮吹仪中以40℃温度吹干。(6) 衍生化:在安瓿瓶中加入50 μL BSTFA+TMCS(99∶1)衍生化试剂,震荡摇匀,再用火焰封闭安瓶,置于70℃烘箱中加热30 min,目标物进行衍生化反应。(7) 准备进样瓶:待冷却后打开安瓿瓶,内容物移入200 μL内插管的2 mL进样瓶中,进行GC-MS分析。
2.4方法学考察
2.4.1线性关系考察制备5个离心管,进行编号,分别加入2 mL空白血液或尿液,每一个离心管中加入100 μL内标丙基解痉素(SKF525a)。在1 ng/mL的标准品储备液中,分别移取100、200、400、800 μL,按顺序放置于离心管中。每个离心管中标准品浓度为50、100、200、400 ng/μL。以待测物浓度为横坐标,待测物与内标物的峰面积比值为纵坐标作图,用加权最小二乘法进行回归运算,求得直线回归方程、线性范围及最低检出限(LOD)。6种毒品及代谢物的标准工作曲线、线性范围、相关系数,见表1。

表1 血液和尿液中药物回归方程和最低检出限
2.4.2回收率、准确度和精密度适量取6种目标毒品标准溶液及内标,加添在2.0 mL空白尿液或空白血液中,配制低、中、高浓度(分别为25、50、200 ng/mL)的血液或尿液的待测溶液,每一种待测溶液各3份,根据工作标准溶液曲线计算个目标毒品溶液的浓度,以待测溶液测得浓度与添加浓度之比求得相对回收率,计算准确度和精密度。6种目标药物的加内标平均回收率为95.9%~105%,RSD为2.13%~5.59%,见表2。
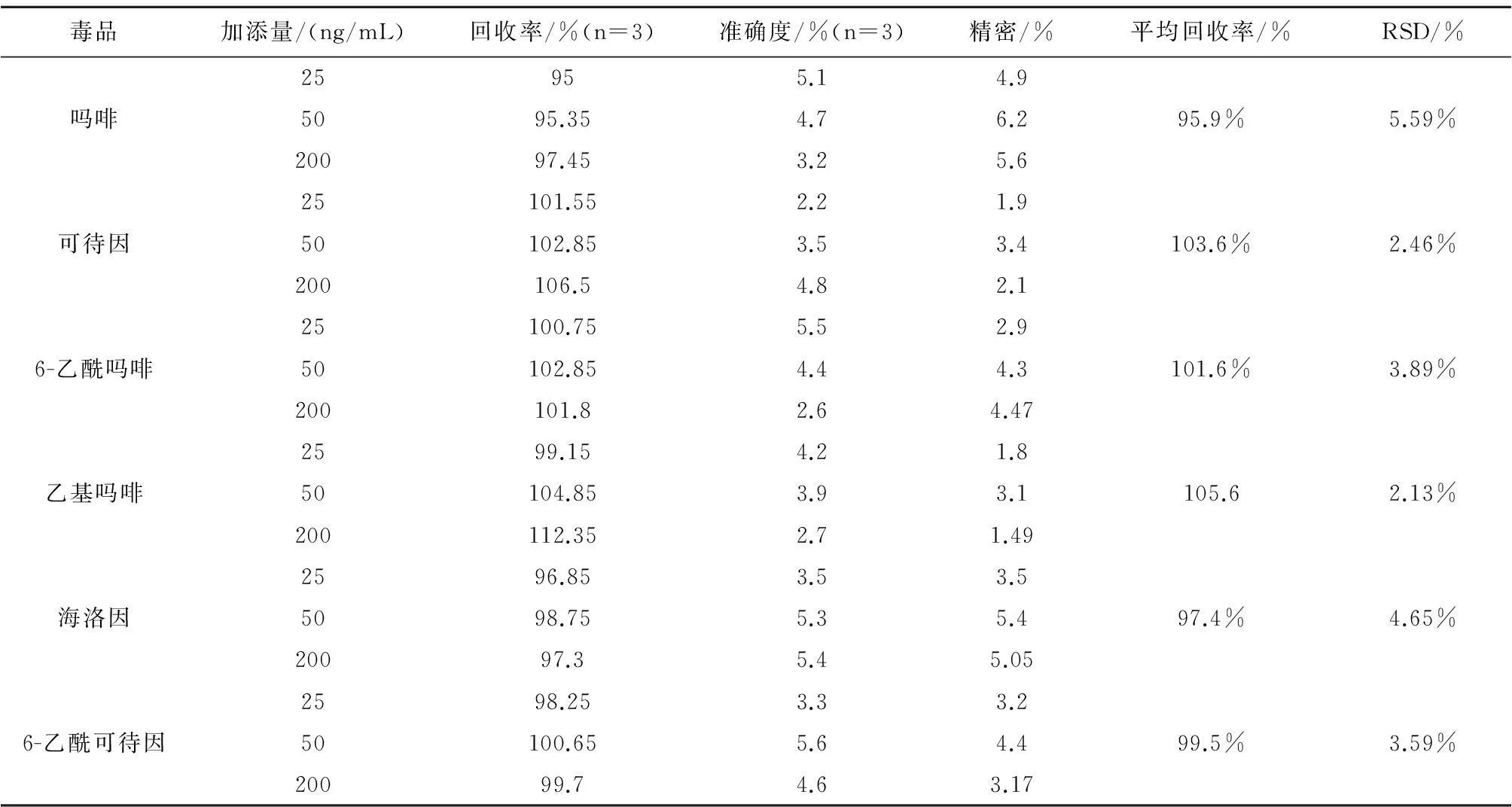
表2 生物样品中阿片类毒品的回收率及相对误差
2.5衍生化衍生化方法种类多,一般常用的有硅烷化、乙酰化、烷基化。阿片类毒品衍生化反应中前2个衍生化反应比较容易,硅烷化是使用硅烷化试剂将待测物分子中的活泼氢用甲基硅烷基取代[8-9]。为了提高对待测物检测灵敏度,本实验利用BSTFA+TMCS(99∶1)衍生化试剂,对待测物混合液进行衍生化,采用GS-MS技术检测,得到了较为满意的的分析结果。相同的质谱条件下,采用全扫描模式(full scan)对标准品待测物和衍生化后的标准品待测物进行检测对比,结果显示质谱灵敏度明显提高。发现衍生化标准品中有6种标准品和衍生化试剂发生反应,其他标准品(杜冷丁、芬太尼、美沙酮)不发生反应。衍生化前后的标准品混合液质谱图见图1、2。

图1 标准品混合液质谱图
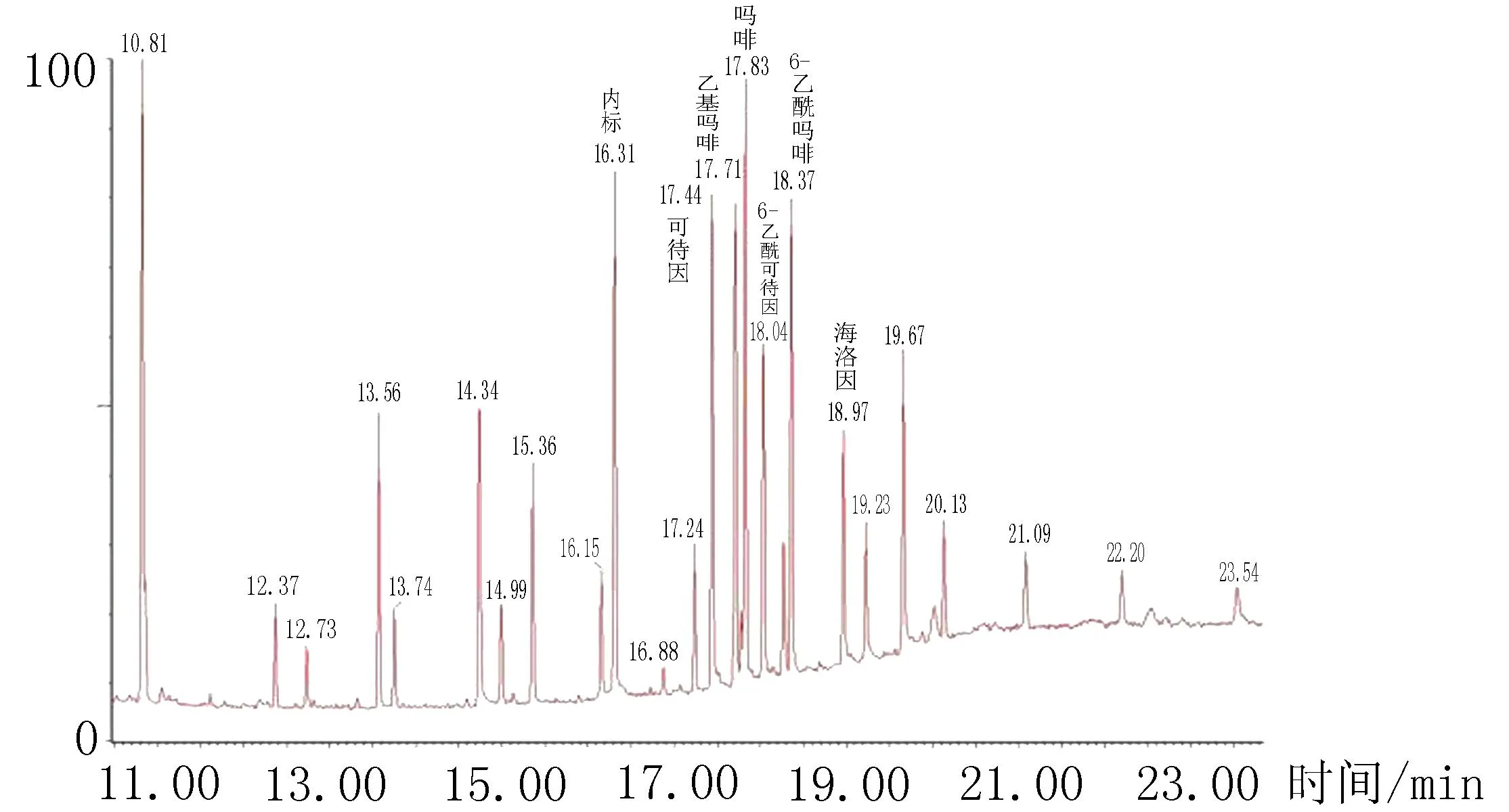
图2 衍生化后的标准品混合液质谱图
2.6目标药物的定性和定量运用气相色谱-联质谱(GC-MS)全扫描模式(Full Scan)和离子选择扫描模式(SIM Scan)对待测毒品及其代谢物进行分析。一般应选择具有较大质荷比(m/z)的一级离子碎片作为母离子,再选2个二级碎片作为子离子,以保证分析的选择性和灵敏度,同时结合保留时间进行分析。本方法的定性结果可靠,完全可以对可疑样品进行确证分析。为避免定量过程中出现的误差,首先采用全扫描模式(Full Scan)获得待测物的特征离子,选择了3个特征离子,用离子选择扫描模式(SIM Scan)通过优化碰撞能量获得产物离子,其中选择了较大质荷比(m/z),以峰面积与对应的浓度进行线性回归,制作标准曲线进行定量,见表3。
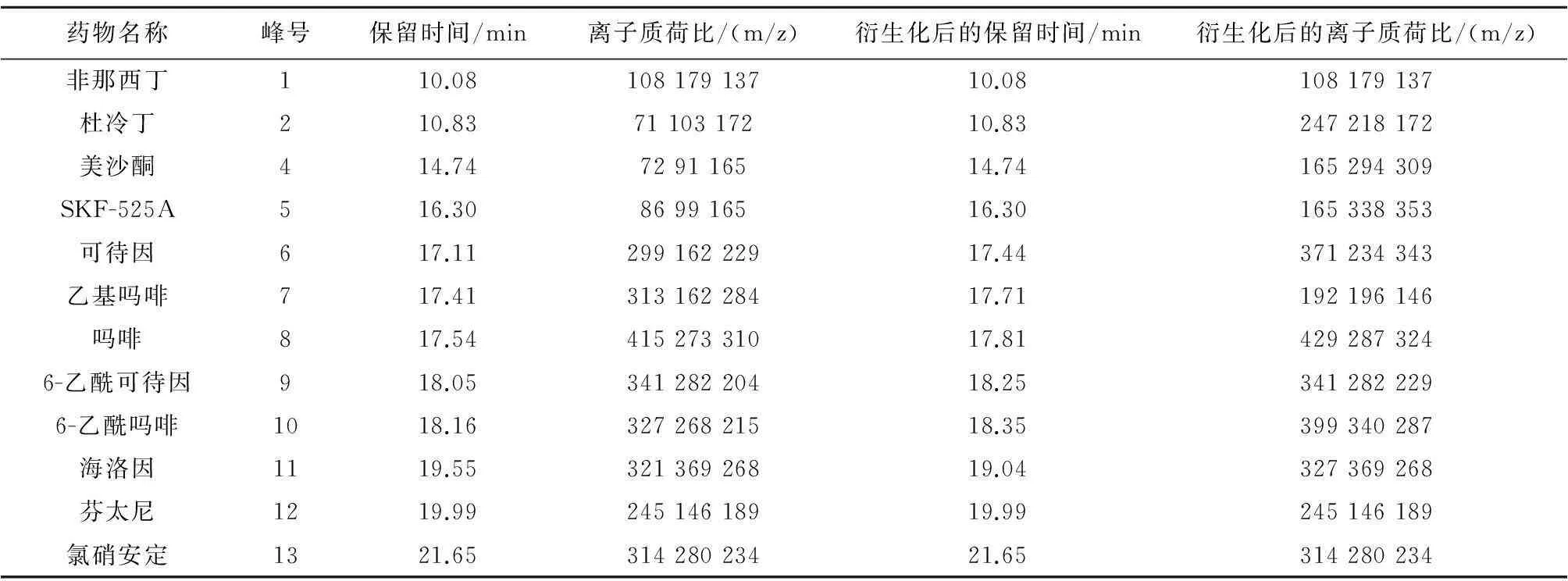
表3 混合样本中的毒品及其代谢物的质荷比
2.7案件样品的含量测定2015年1月,乌鲁木齐市某派出所发现一具尸体,死者男,怀疑为吸毒死亡,尿检呈吗啡类毒品阳性反应。派出所送来死者的尿液约20 mL、血液10 mL要求进行实验室确证检测。尿液通过已建立的前处理方法,分离、提取后,采用常规气相色谱-联质谱仪(GC-MS)全扫描模式进行检测,尿液检测出可待因、吗啡成分、6-乙酰吗啡成分,可待因含量>400 ng/mL,吗啡为384 ng/mL,6-乙酰吗啡为278 ng/mL,血液检测量极小,确证了吸食海洛因的违法事实。死者尿液中检出的3种目标药物的SIM 模式总离子流图,见图3、表4。
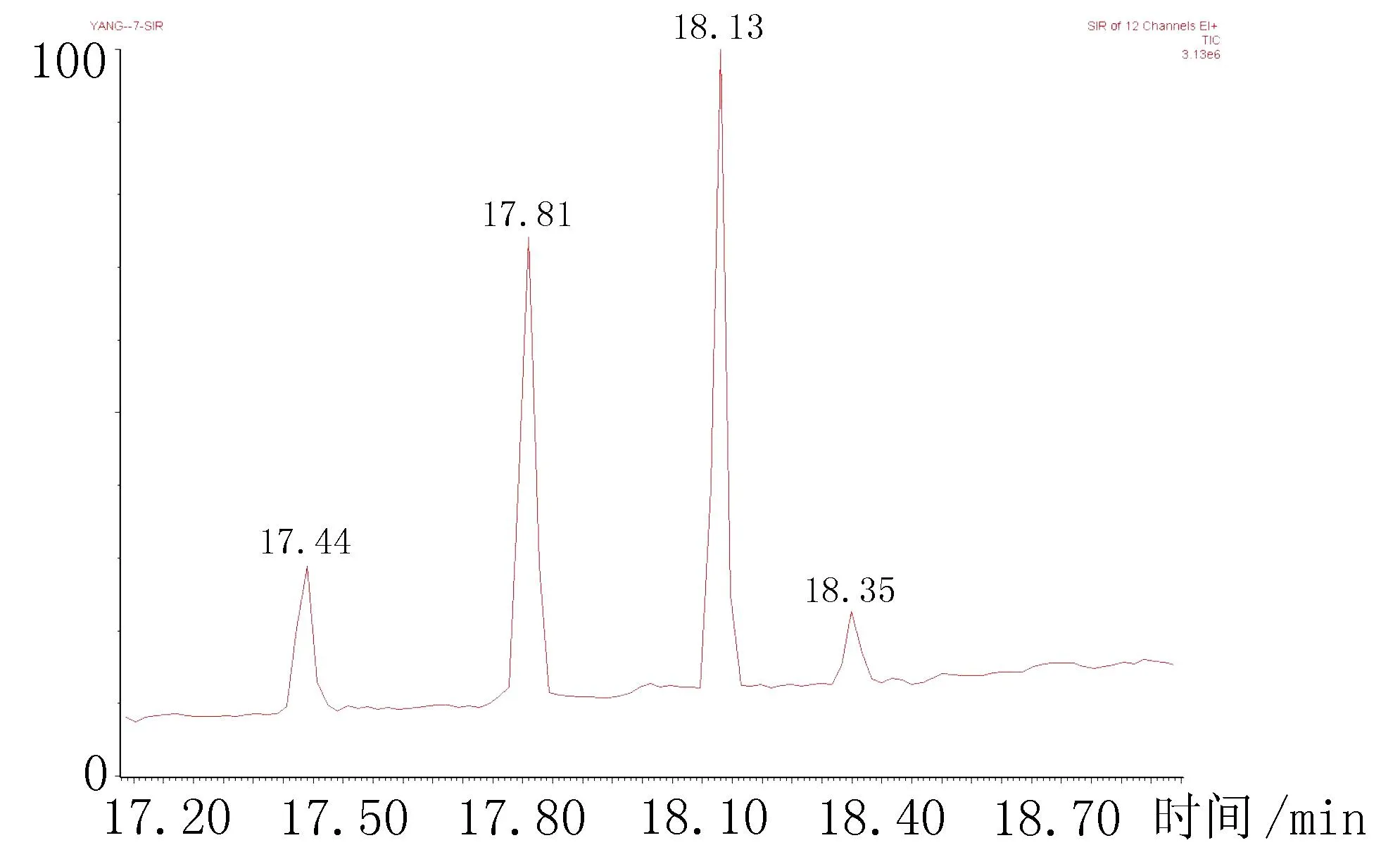
图3 死者尿液中检出的3种目标药物的SIM模式总离子流图
表4检尿中3种目标药物的SIM模式质荷比及保留时间/min

待测物保留时间质荷比/(m/z)可待因17.44371234343吗啡17.814292873246-乙酰吗啡18.36399340287
3讨论
3.1质谱条件的优化阿片类药物是分子量>500的极性分子,因而选择了弱极性毛细管柱子,本实验所拥有的气相色谱-质谱联用仪(GC-MS),选择了弱极性毛细管柱子(DB-5MS),采用全扫描(Full Scan)方式和选择离子质荷比(Select Ion Monitoring,SIM)方式进行测试,将待测物的保留时间与结合特征离子质谱图进行对比定性分析。选择离子质荷比模式检测灵敏度更高,可做定量分析,对每一种标准品选择了3个(m/z)特征离子,根据3个(m/z)特征离子在同一个保留时间出现的峰重叠性来定性标准品,该模式不但可以降低混合物质的影响,而且在扫描速度不变的情况下,增加了对特定离子的扫描次数,提高了扫描准确度和精密度,大幅度提高了检测灵敏度。本实验对几种程序升温方式进行了比较,建立的分析条件能使混合阿片类药物很好地分离。经过优化,得到了较为理想的分析色谱图[10]。
3.2固相萃取条件固相萃取法(SPE)一般用于复杂生物样品的处理,例如血液、血浆、生物体液中的酸性、中性或者碱性毒品萃取。为确保对较多毒品类型均有效,将分析方法简单化,并提高检测灵敏度,本实验将Bond Elut Plexa固相萃取小柱应用于常规药(毒)物的前处理,结果较为满意。因运用气相色谱-联质谱(GC-MS)检测预处理过的毒品,混杂物质对检测效果的影响比较小,得到了满意的检测结果。
本研究建立了一种基于固相萃取SPE法与衍生化技术,对生物样样品进行前处理,利用GC-MS法对生物样品(血液、尿液)中的阿片类毒品及其代谢物进行定性、定量检测。该方法灵敏度高,适用于阿片类毒品滥用者的尿样、血样在实验室常规检测。
参考文献:
[1]杨良.药物依赖学[M].北京:人民卫生出版社,2015:265-275.
[2]孟品佳,王继芬,王燕燕,等.鸦片类毒品酰化与硅烷化在GC-MS分析中的比较[J].化学试剂,2010,32(1):69-73.
[3]Ghazi-Khansari M, Zendehdel R, PiralihamedaniI M, et al. Determination of morphine in the plasma of addicts in using Zeolite Y extraction following high Performance liquid chromatography[J].Clin Chin Acta,2006,364:235-238.
[4]Pujadas M,Pichini S,Civit E, et al. A simple and reliable procedure for the determination of psychoactive drugs in oral fluid by gas chromatography-mass spectrometry[J]. Pharm Biomed Anal,2007,44:594-601.
[5]Lopez P, Bermejo AM,Tabernero MJ, et al. Determination of cocaine and heroin with their respective metabolites in meconium by gaschromatography-mass spectrometry[J]. Appl Toxicol,2007,27:464-471.
[6]Guillot E,Mazancourt PD,Durigon M, et al. Morphine and 6-acetylmorphine concentrations in blood,brain,spinal cord,bone marrow and bone after lethal acute or chronic diacetylmorphine administration to mice[J].Forensic Sci lnt,2007,166:139-144.
[7]Meng PJ. Qualitative and quantitative analysis of ecatacy with combined derivatization of MSTFA and MBTFA[J].China Lab,2000,19:90-92.
[8]魏万里.SPE-GC-MS/MS-SRM法快速检测尿液中12种常见毒品成分及代谢物[J].中国人民公安大学学报,2012,3:27-32.
[9]邓艳萍,孙桂宽,魏伟,等.尿样中滥用药物的检测分析[J].中国药物依赖性杂志,2002,11(2):117-119.
[10]王燕燕,孟品佳,等.SPE-GC/MS法检测血液中的鸦片类毒品[J].应用化学,2009,26(12):1496-1498.
(本文编辑施洋)
Qualitative and quantitative detection of opioid drugs and their metabolites in the blood and urine by GC and GC-MS methods
Nurali Tayir1, MAO Xinmin2, CHE Wensen3, Askar Arken1, Akbar Rahman1
(1Department of Forensic Medicine, School of Preclinical Medicine;2College of Traditional Chinese Medicine, Xinjiang Medical University, Urumqi 830011, China;3Urumqi Public Security Bureau, Urumqi 830000, China)
Abstract:ObjectiveEstablish gas chromatography (GC) and gas-mass spectrometry (GC-MS) methods to detect opioid drugs and their metabolites in the blood and urine. MethodsAdded pH6 phosphate buffer solution into urine or blood sample as 0.01 M to adjust pH at 5-6, brought it into solid phase extraction column (SPE), rinsed the column by deionized water and 0.01 M HCl, and then washed SPE column with CH3OH. Extracted by CH2Cl2/CH3CH2OHCH3 mixture which were contained 2% ammonia and dried up, then derivatized with BSTFA+TMCS (99∶1) reagent, took SKF-525 as internal standard, tested the content of the sample by DB-5 MS chromatographic column at ion (m/z) scan mode (SIM) and the full scan model of GC and GC-MS. ResultsThere were good linear relationship between the contents of drugs (heroin, morphine, codeine, 6-acetyl codeine, 6-acetyl morphine, and ethyl morphine) and peak area in the range of 25~400 μg/L, in which the correlation coefficient r was bigger than 0.9907; the average recovery rate of internal standard was 95.9~101.6%; the relative standard error (RSD) was 2.13%; the minimum limit of detection (LOD) was 1 μg/L, the minimum limit of quantitative (LOQ) was 25 μg/L. Codeine, morphine and 6-acetyl morphine in case urine sample could be detected. The content of codeine was >400 μg/L, morphine was 384 μg/L, and 6-acetyl morphine was 278 μg/L, while detection amount of blood samples were extremely low, but the illegal facts of heroin could be confirmed. ConclusionThe methods were simple, rapid, high sensitivity and accurate quantitative and qualitative, which will be suitable for the screening and confirming test of blood and urine samples of drug addicters or drug abusers.
Keywords:opioid drugs; GC-MS; The SPE extraction method; Derivatization; Heroin
基金项目:新疆维吾尔自治区自然科学基金(201133132); 新疆维吾尔自治区乌鲁木齐市科技计划项目(Y111310019)
作者简介:努尔艾力·塔依尔(1989-),男(维吾尔族),在读硕士,研究方向:法医毒物分析学研究。 通信作者:艾克拜尔·热合曼,男(维吾尔族),博士,副教授,硕士生导师,研究方向:法医毒物分析学、新药研发,E-mail: 1050558470@qq.com。
中图分类号:R331
文献标识码:A
文章编号:1009-5551(2016)07-0849-06
doi:10.3969/j.issn.1009-5551.2016.07.009
[收稿日期:2016-3-6]
