氮杂环酰胺化合物合成研究
2016-06-06高建荣李郁锦浙江工业大学化学工程学院浙江杭州310014
沈 超,张 侃,高建荣,李郁锦(浙江工业大学 化学工程学院,浙江 杭州 310014)
氮杂环酰胺化合物合成研究
沈超,张侃,高建荣,李郁锦*
(浙江工业大学化学工程学院,浙江杭州310014)
摘要:对氮杂环酰胺化合物的合成方法进行了研究:在四氢呋喃溶剂中,在催化剂4-二甲氨基吡啶存在下,以苯并噻唑胺、噻二唑胺等杂环胺和4-取代苯甲酰氯为原料,经酰胺化反应,合成了12个氮杂环酰胺化合物。产率高达83.1%~96.3%。该法原料易得、操作简单、条件温和、收率高、底物适用范围广。
关键词:氮杂环酰胺;苯并噻唑;1,3,4-噻二唑;4-二甲氨基吡啶
酰胺类化合物是广泛应用的化工原料,也是合成染料、色素、农用化学品及医药品的重要中间体[1];且其结构极易与有机体形成氢键,从而显示出良好的药物活性[2-3]。含苯并噻唑、1,3,4-噻二唑结构单元的氮杂环酰胺化合物是一类重要的化合物,在目前新农药研发中,此类化合物作为杀菌剂已得到广泛应用[4]。除了杀菌活性,很多酰胺类化合物还呈现出独特的杀虫机制和良好的除草性能,因此对于酰胺类化合物的合成研究具有十分广阔的发展前景[5]。
酰胺传统的合成方法主要是由羧酸及其衍生物与胺发生亲核取代反应得到。除此之外,酰胺的合成还可由Schmidt反应、Staudinger ligation反应、Beckmann重排、腈基部分水解得到[6]。文献报道氮杂环酰胺化合物的合成主要采用传统酰胺合成法,如羧酸与胺缩合法[5]、氰基转化法[7]、酰氯法[8]等。本文以取代2-氨基苯并噻唑、2-氨基-5-苯基-1,3,4-噻二唑,色胺等胺和4-取代苯甲酰氯为原料,研究酰氯与胺的酰胺化反应条件,最终得到一条操作简单、后处理方便、条件温和、收率高、底物适用范围广的酰胺合成路线。
1 实验部分
1.1仪器与试剂
1H NMR和13C NMR在Bruker -AV -500 (500 MHz)核磁共振仪上测定,溶剂为DMSO-d6和CDCl3,内标为四甲基硅烷(TMS)。熔点测定采用X-4数显显微熔点仪(温度计未校正);TLC板用硅胶GF-254(青岛海洋化工制)。
实验所用化学试剂和溶剂为市场采购分析纯级别的试剂,除了论文中有特别说明以外,没有进一步纯化。
1.2N-取代苯甲酰胺衍生物的合成方法
1.2.1N-苯并噻唑基苯甲酰胺衍生物的合成
N-苯并噻唑基苯甲酰胺衍生物(3a~3f)的合成通法如下,以3a为例:
在50 mL单口瓶中依次加入2-氨基苯并噻唑(0.3004 g,2 mmol),三乙胺(0.2021 g,2 mmol),DMAP(0.0122 g,0.1 mmol),四氢呋喃30 mL,室温下逐滴滴加苯甲酰氯(0.3373 g,2.4 mmol)。TLC检测原料噻唑反应完全(2 h),加入30% Na2CO3溶液5 mL,搅拌10 min,加水150 mL稀释,有大量白色固体析出。抽滤,用水洗涤产品,干燥称重。得到目标产品3a(0.4705 g,产率为92.5%)。
3a:白色固体,产率92.5%,m.p.183℃~184℃(ref:185℃~187℃)[9];1H NMR(500 MHz,CDCl3):δ 11.43(s,1H),8.01(d,J=7.4 Hz,2H),7.87 (dd,J=5.9,2.9 Hz,1H),7.59(t,J=7.4 Hz,1H),7.45(t,J=7.8 Hz,2H),7.34-7.29(m,3H)。
3b:白色固体,产率96.3%,m.p.178℃~179℃(ref:180℃~181℃)[9];1H NMR(500 MHz,CDCl3):δ 11.65(s,1H),7.90(d,J=8.2 Hz,2H),7.86 (d,J=7.3 Hz,1H),7.29(m,3H),7.21(d,J=8.0 Hz,2H),2.39(s,3H)。
3c:白色固体,产率91.4%,m.p.184℃~185℃(ref:185℃~186℃)[9];1H NMR(500 MHz,CDCl3):δ 11.21(s,1H),7.95~7.92(m,2H),7.90~7.87 (m,1H),7.43(dt,J=6.9,2.0 Hz,3H),7.39~7.33 (m,2H)。
3d:白色固体,产率93.1%,m.p.189℃~190℃(ref:190℃~192℃)[10];1H NMR(500 MHz,CDCl3):δ 11.29(s,1H),7.98(d,J=8.8 Hz,2H),7.88~7.84 (m,1H),7.45~7.41(m,1H),7.35~7.28(m,2H),6.91(d,J=8.7 Hz,2H),3.84(s,3H)。
3e:黄色固体,产率95.4%,m.p.293℃~294℃(ref:296℃~296.5℃)[10];1H NMR(500 MHz,DMSO-d6):δ 13.28(s,1H),8.38~8.34(m,4H),8.02 (d,J=7.8 Hz,1H),7.78(d,J=8.0 Hz,1H),7.51~7.46(m,1H),7.36(dd,J=11.2,3.8 Hz,1H)。
3f:白色固体,产率90.8%,m.p.199℃~200℃(ref:200℃~202℃)[11];1H NMR(500 MHz,DMSO-d6):δ 11.58(s,1H),8.00(d,J=7.3 Hz,2H),7.57 (t,J=7.4 Hz,1H),7.44(t,J=7.8 Hz,2H),7.32 (d,J=2.5 Hz,1H),7.13(d,J=8.9 Hz,1H),6.87 (dd,J=8.9,2.5 Hz,1H),3.88(s,3H)。
1.2.2N-(5-苯基-1,3,4-噻二唑-2-基)苯甲酰胺衍生物的合成
该反应起始原料为取代苯甲酸,参考文献[12]的合成方法,得到2-氨基-5-苯基-1,3,4-噻二唑及2-氨基-5-(4-甲氧基苯基)-1,3,4-噻二唑,而后再合成酰胺。实验步骤如下:
在25 mL两口瓶中依次加入苯甲酸(0.2442 g,2 mmol),氨基硫脲(0.1823 g,2 mmol),搅拌下分三次缓慢注射三氯氧磷1.2 mL,80℃反应2.5 h。反应完毕,冷却至室温,搅拌下分三次注射2.5 mL水,110℃反应4 h。TLC检测反应完全,冷却至室温,将反应液倒入15 mL冰水中,用40%NaOH溶液中和至pH=8。有大量白色沉淀产生,过滤,用水洗涤,滤饼用乙醇重结晶,得到产品2-氨基-5-苯基-1,3,4-噻二唑(0.3130 g,产率为88.3%)。
2-氨基-5-苯基-1,3,4-噻二唑:白色固体,产率88.3%,m.p.224℃~226℃(ref:220℃)[12];1H NMR(500 MHz,CDCl3):δ 7.82(dd,J=6.5,3.1 Hz,2H),7.45(dd,J=5.0,1.8 Hz,3H),5.21 (s,2H)。
2-氨基-5-(4-甲氧基苯基)-1,3,4-噻二唑:白色固体,产率86.4%,m.p.185℃~187℃(ref:181℃)[12];1H NMR(500 MHz,DMSO-d6):δ 7.69 (d,J= 8.6 Hz,2H),7.30(s,2H),7.02(d,J= 8.6 Hz,2H),3.80(s,3H)。
N-(5-苯基-1,3,4-噻二唑-2-基)苯甲酰胺衍生物(3g~3j)的合成通法如下,以3g为例:
在50 mL单口瓶中依次加入2-氨基-5-苯基-1,3,4-噻二唑(0.3544 g,2 mmol),三乙胺(0.2021 g,2 mmol),DMAP(0.0122 g,0.1 mmol),四氢呋喃30 mL,室温下逐滴滴加苯甲酰氯(0.3373 g,2.4 mmol)。TLC检测原料噻唑反应完全(2.5h)。加入30%Na2CO3溶液5mL,搅拌10min,加水150 mL稀释,有大量白色固体析出。抽滤,用水洗涤产品后乙醇重结晶得纯品3g(0.4861 g,产率为86.4%)。
3g:白色固体,产率86.4%,m.p.234℃~235℃(ref:237℃~238℃)[13];1H NMR(500 MHz,CDCl3):δ 12.08(s,1H),8.30~8.28(m,2H),7.99 (dd,J=6.5,3.1 Hz,2H),7.70(t,J=7.4 Hz,1H),7.60(t,J=7.7 Hz,2H),7.54~7.51(m,3H)。
3h:白色固体,产率84.3%,m.p.249℃~251℃(ref:245℃)[13];1H NMR(500 MHz,DMSO-d6):δ 13.00(s,1H),8.17(d,J=8.7 Hz,2H),8.03~7.94 (m,2H),7.55(d,J=5.7 Hz,3H),7.11(d,J=8.7 Hz,2H),3.87(s,3H)。
3i:黄色固体,产率83.1%,m.p.>300℃(ref:271℃)[14];1H NMR(500 MHz,DMSO-d6):δ 13.65 (s,1H),8.38(dd,J=17.1,8.4 Hz,4H),8.00(d,J= 5.4 Hz,2H),7.56(d,J=5.3 Hz,3H).
3j:米色固体,产率85.5%,m.p.230℃~232℃;1H NMR(500 MHz,DMSO-d6):δ 13.09(s,1H),8.15(d,J=7.5 Hz,2H),7.93(d,J=8.7 Hz,2H),7.68(t,J=7.3 Hz,1H),7.58(t,J=7.6 Hz,2H),7.11(d,J= 8.7 Hz,2H),3.85(s,3H)。
1.3 N1,N2-二取代乙二酰胺化合物的合成反应
1.3.1N1,N2-二(苯并噻唑-2-基)乙二酰胺的合成
在50 mL单口瓶中依次加入2-氨基苯并噻唑(0.3004 g,2 mmol),三乙胺(0.2021 g,2 mmol),DMAP(0.0122 g,0.1 mmol),四氢呋喃30 mL,室温下逐滴滴加乙酰氯(0.3373 g,1.2 mmol)。TLC检测原料噻唑反应完全(2 h)。加入30%Na2CO3溶液5 mL,搅拌10 min,加水150 mL稀释,有大量黄色固体析出。抽滤,用水洗涤产品。干燥称重。得目标产品6a(0.6542 g,产率为92.3%)。
6a:黄色固体,产率92.3%,m.p.350℃~351℃;1H NMR(500 MHz,DMSO-d6):δ 12.76(s,2H),8.15(d,J= 6.8 Hz,2H),7.95(d,J=7.6 Hz, 1H),7.74(d,J=8.0 Hz,1H),7.45~7.38(m,1H),7.32~7.25(m,1H),6.74(d,J=6.7 Hz,2H)。
1.3.2N1,N2-二色氨基乙二酰胺的合成
在50 mL单口瓶中依次加入色氨(0.3204 g,2 mmol),三乙胺(0.2021 g,2 mmol),DMAP (0.0122 g,0.1 mmol),四氢呋喃30 mL,室温下逐滴滴加乙酰氯(0.3373 g,1.2 mmol),TLC检测原料反应完全(2 h)。加入30%Na2CO3溶液5 mL,搅拌10 min,加水150 mL稀释,有大量黄色固体析出。抽滤,用水洗涤产品。干燥称重。得目标产品6b(0.7180 g,产率为95.9%)。
6b:黄色固体,产率95.9%,m.p.343℃~344℃(ref:>320℃)[15];1H NMR(500 MHz,DMSO-d6):δ 10.83(s,1H),8.85(t,J=6.1 Hz,1H),7.58(d,J= 7.9 Hz,1H),7.34(d,J=8.1 Hz,1H),7.17(d,J=2.2 Hz,1H),7.11~7.03(m,1H),7.01~6.95(m,1H),3.44(dd,J=14.7,6.6 Hz,2H),2.89(t,J=7.5 Hz,2H)。
2 结果与讨论
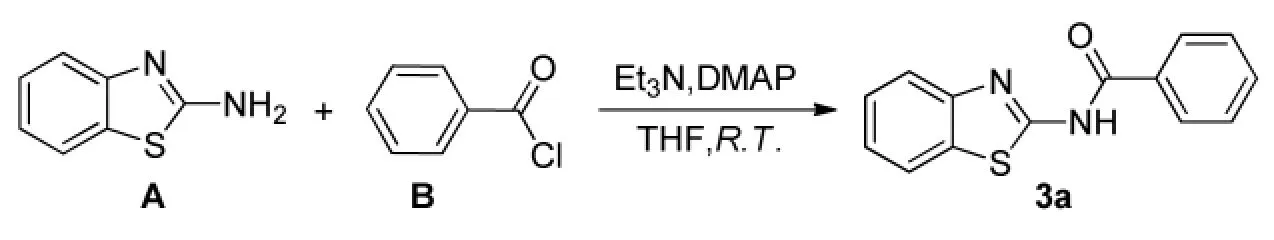
图1 3a合成路线Fig.1 Synthetic route of 3a
本文以2-氨基苯并噻唑和苯甲酰氯为起始原料,以3a为例对合成路线进行探索。首先对催化剂进行选择。不采用催化剂,以二氯甲烷作溶剂,三乙胺作缚酸剂,加热回流反应4 h剩余大量原料,产率低于40%;采用DMAP(摩尔量5%)作为催化剂,以同样的条件在室温下反应,产率即可达到72%。其次,对溶剂进行了筛选,在二氯甲烷和甲苯中产物粗品产率可达到71%,但粗品需要过柱提纯,步骤繁琐且收率不高。而在THF中,反应产生的三乙胺盐酸盐析出,促进反应向正方向进行,转化率高;且加水产物即可析出,提纯过程方便,产率可达90%。再次,对碱进行了筛选,发现在THF中用无机碱碳酸钠或氢氧化钠产率小于50%,反应较长时间原料不能反应完全,而用有机碱三乙胺或N,N-二异丙基乙胺,产率均能达到90%以上。实验结果表明,该反应均相体系效果更佳。然后对加料方式也进行了筛选,发现以酰氯滴加的方式优于三乙胺滴加。若以三乙胺滴加,酰氯和胺直接混合,反应液中有明显副产物产生。若以酰氯滴加,反应温和,无明显的副产物产生,选择性好,产率增加。本文对投料比进行了筛选,尝试用胺与酰氯摩尔比=1∶1,1∶1.1,1∶1.2~1∶2投料,发现酰氯小于1.2当量时,同样条件下胺反应不完全;酰氯大于1.2当量时,最终产品含有较多酰氯,纯度达不到要求。故选择最佳摩尔比为1∶1.2。合成路线见图1。
在此基础上,本文以2-氨基苯并噻唑为底物胺,在四氢呋喃中,在催化剂DMAP存在下,室温下先将氨和缚酸剂三乙胺在四氢呋喃中搅拌5 min,后滴加取代苯甲酰氯,反应2 h,获得90%以上的收率合成目标酰胺化合物,且通过TLC跟踪反应,无明显副产物生成;该方法后处理简单,只需将反应液用少量碳酸钠溶液洗涤(除去酰氯分解产生的酸),倒入适量水中,过滤水洗即可得到纯净的目标产物。反应合成路线见图2。实验结果见表1。

图2 N-取代苯甲酰胺衍生物合成路线Fig.2 Synthetic route of N-substituted benzamide derivatives
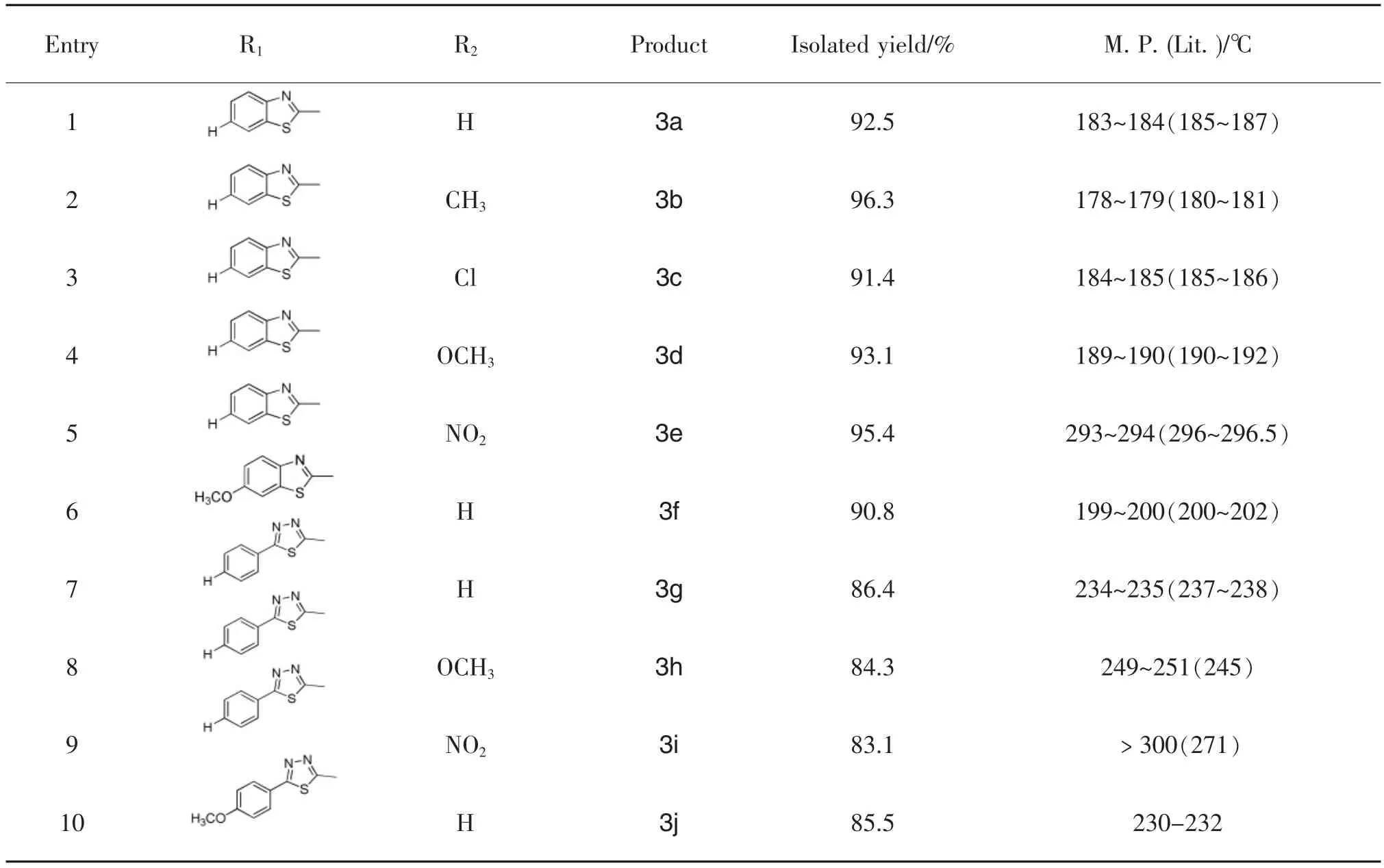
表1 N-取代苯甲酰胺衍生物的合成反应Table 1 Synthesis of N-substituted benzamide derivatives
在探索的最佳条件下,笔者合成了6个N-苯并噻唑基苯甲酰胺衍生物,包括了酰氯苯环上给电子取代基甲基、甲氧基,吸电子取代基硝基,卤原子等基团,以及苯并噻唑6位上甲氧基取代基。这些化合物均达到了90%以上的收率,其中以酰氯苯环上甲基取代收率最高,达到了96.3%。
本文对拓展反应底物也进行了研究。尝试了以2-氨基-5-苯基-1,3,4-噻二唑化合物为原料合成,通常含苯并噻唑,1,3,4-噻二唑结构单元的化合物具有较好的生物活性。但因其溶解性的关系,以及氨基的反应活性较脂肪族胺和苯胺差,且往往难于达到较理想的转化率。表1实验结果表明:按照以上合成条件,对溶解性较差的2-氨基-5-苯基-1,3,4-噻二唑反应效果亦显著。收率普遍达到83%以上,最高可以达到86.4%。该路线与文献[20]报道的方法相比,起始原料易得,反应条件温和,收率更高,且不必经过剧毒中间体异硫氰酸酯。因此,本文所报道的方法,在氮杂环酰胺的合成中优势尤为明显,且该反应底物的适用范围宽。
接着,本文对反应底物做了进一步拓展,采用较活泼的伯胺—色胺和不活泼的杂环胺—2-氨基苯并噻唑两种原料合成两种双酰胺衍生物。合成路线见图3。

图3 N1, N2-二取代乙二酰胺化合物合成路线Fig.3 Synthetic route of N1, N2-disubstituted double amide derivatives

表2 N1, N2-二取代乙二酰胺化合物的合成反应Table 2 Synthesis of N1, N2-disubstituted double amide derivatives
实验结果显示:该路线对双酰胺化反应亦适用,效果均显著,达到了92%以上的收率,其中使用活泼胺产率更高,达到了95.9%,进一步展现了该方法的优势。实验结果见表2,所有产物结构都经熔点测试和1H NMR表征。
3 结论
综合考虑各种因素后得出结论:在THF溶剂中,在催化剂DMAP存在下,n(胺):n(酰氯)=1∶1.2,以酰氯滴加的方式加料,室温下反应1~2 h,可获得90%以上的产率得到N-苯并噻唑基苯甲酰胺衍生物。并对该方法的适用范围进行拓展,同样以85%和92%的高产率分别合成了N-(5-苯基-1,3,4-噻二唑-2-基)苯甲酰胺衍生物、N1,N2-二取代乙二酰胺衍生物。此方法原料易得、操作简单、后处理方便、条件温和、收率高、底物适用范围广,对于氨基的亲核进攻活性相对较小的杂环胺反应效果均显著。它提供了一种广泛合成有机合成中间体酰胺的新方法。
参考文献:
[1]Mahran M A,El-nassry S M F,Allam S R.Synthesis of some new benzothiazole derivatives as potential antimicrobial and antiparasotic agents[J].Pharmazie,2003,58(8):527-530.
[2]Sawada Y,Yanai T,Nakagawa H,et al.Synthesis and insecticidal activity of benzoheterocyclic analogues of N’-benzoyl-N-(tert-butyl)benzohydrazide[J].Pest ManagementScience,2003,59:25-35.
[3]El-Gaby M A,Micky J,Taha N,et al.Antimicrobial activity of some novel thiourea,hydrazine,fused pyrimidine and 2-(4-substituted)anilino benzoazole derivatives containing sulfonamide moieties[J].Journal of the Chinese Chemical Society,2002,49:407-414.
[4]王振军,刘斌,赵卫光,等.1-取代-1H-1,2,3-三唑-4-甲酰胺类化合物的设计合成与生物活性[J].有机化学,2011,31(3):317-323.
[5]刘晓,陈万里,莫为民.2-碘苯甲酰胺类化合物的合成[J].浙江化工,2014,4(8):11-13.
[6]盛国柱,张炜.酰胺官能团构建方法研究新进展[J].有机化学,2013,33:2271-2282.
[7]赵红伟,梁诚.2,6-二氟苯甲酰胺合成及下游农药开发研究[J].有机氟工业,2010,2:28-34.
[8]郑玉国,周莉,薛伟,等.新型双酰胺衍生物的合成及抗癌活性[J].精细化工,2012,29(3):285-289.
[9]Al-Nima H H,Al-Aulaymi J R,Hashim O K.Reaction of 2-aminobenzothiazole and 3-amino-5-methylisoxazole with isothiocyanates[J].Journal of the Iraqi chemical Society,1986,11:13-24.
[10]Augustin M,Richter M,Salas S.Reactions with N-acylimino dithiocarbonic acid diesters[J].Journal fuer Praktische,1980,322:55-68.
[11]Yuan J F,Zhang M Z.Synthesis of some 2-substituted aminobenzothiazoles[J].Beijing Daxue Xuebao,Ziran Kexueban,1988,24:504-506.
[12]刘玉婷,周英,尹大伟.2-氨基-5-芳基-1,3,4-噻二唑的合成[J].精细石油化工,2011,28(1):63-63.
[13]Neidlein,Richard,Tauber,J.Synthesis of(N -acylamino)-1,3,4-thiadiazoles[J].Pharma -zeutische Zentralhalle fur Deutschland,1968,107:430-432.
[14]李英俊,丁万刚,靳焜,等.2-芳基-5-(4-硝基苯甲酰氨基)-1,3,4-噻二唑的合成[J].有机化学,2009,29(1):82-88.
[15]Jiang W Q,Wen R,Jean Y L.β-Carbolines.1.synthesis of several new bis-β-carboline compounds[J].Jurnal Chinese Pharmaceutical Seienees,1999,8(3):177-179.
修回日期:2016-01-08
Synthesis of Nitrogen Heterocycle-Containing Amide Derivatives
SHEN Chao,ZHANG Kan,GAO Jian-rong,LI Yu-jin*
(College of Chemical Engineering,Zhejiang University of Technology,Hangzhou 310014,Zhejiang,China)
Abstract:A study for synthesis of nitrogen neterocycle-containing amide derivatives was provided.In this method,benzothiazolamine or thiadiazole amine reacted with benzoyl chloride in 1:1.2 molar ratio in THF with DMAP as the catalyst to form twelve amide derivatives with 83.1%~96.3%yields.The method was of readily available raw materials,simple operation,mild conditions,high yield,apply a wide range of substrates.
KeyWords:heterocyclic amide;benzothiazole;1,3,4-thiadiazole;DMAP
文章编号:1006-4184(2016)5-0005-05
作者简介:沈超(1990-),男,硕士研究生。
*通讯作者:李郁锦,女,副教授。E-mail:lyjzjut@zjut.edu.cn。
