缅甸蟒养殖种群的遗传多样性分析
2016-06-04段玉宝王英树马建章白素英戎西南林业大学林学院云南省森林灾害预警与控制重点实验室云南昆明6504东北林业大学野生动物资源学院黑龙江哈尔滨150040上海野生动物园上海0199
段玉宝王英树马建章白素英戎 可(1.西南林业大学林学院,云南省森林灾害预警与控制重点实验室,云南昆明6504;.东北林业大学野生动物资源学院,黑龙江哈尔滨150040;.上海野生动物园,上海0199)
缅甸蟒养殖种群的遗传多样性分析
段玉宝1,2王英树3马建章2白素英2戎 可2
(1.西南林业大学林学院,云南省森林灾害预警与控制重点实验室,云南昆明650224;2.东北林业大学野生动物资源学院,黑龙江哈尔滨150040;3.上海野生动物园,上海201399)
摘要:利用微卫星标记分析缅甸蟒养殖群体的遗传多样性,结果表明:在42个缅甸蟒个体中,共检测到76个等位基因,平均有效等位基因数为5.78个,平均期望杂合度为0.815,平均多态信息含量为0.78,8个微卫星位点呈现高度多态性;仅有3个位点显著偏离了Hardy-Weinberg平衡;缅甸蟒海南养殖群体表现出较高的遗传多样性水平。突变-漂移平衡分析结果表明,群体部分位点杂合显著过剩,并未显著偏离突变-漂移平衡,近期没有经历过瓶颈效应,群体数量无明显下降。
关键词:缅甸蟒;微卫星;遗传多样性;养殖种群
缅甸蟒(Python bivittatus)属于蛇亚目(Serpentes)、蟒科(Pythonidae)、蟒属(Python)蛇类,是我国一级重点保护野生动物,国际《濒危野生动植物国际贸易公约》(CITES)附录Ⅱ物种[1-2]。缅甸蟒主要分布在东南亚各国,以及我国的海南、云南(南部)、广东、广西、福建、香港、四川等地[3-4]。栖息于低海拔的热带、亚热带低山丛林,由于过度人为干扰和破坏,使其栖息地不断萎缩、片段化,其种群状况呈锐减趋势[5]。
缅甸蟒作为模式生物,在生物医学、发育生物学、分子生态学、分子遗传进化等领域中起到关键作用[6-10]。目前,不仅在蟒蛇基因组测序、快速微卫星标记的发展等方面进行了研究,在相关物种间引物的通用性等方面也进行了探讨[11-14]。利用分子生物学手段,Groot等在Artis动物园发现缅甸蟒存在孤雌生殖,然而在野生种群的研究中并没有发现这一现象[15-16]。Collins等从Jordan等报道的27对蟒科蛇类的引物中选取10对,分析了美国大沼泽国家公园(Everglades National Park)入侵的缅甸蟒的种群遗传特征[16-17]。但是在缅甸蟒原始分布地中国,在分子生物学方面研究甚少,王志方等扩增了缅甸蟒的Dmrt基因的DM结构域,You等、柯文灿等、刘雅芳等均利用线粒体DNA做了分子鉴定和遗传分化等方面的研究[18-21]。
微卫星DNA因其分布广泛、变异度极高和易于检测等特点能清楚地了解种群间关系、种群动态及其进化历史,从而为野生动物的繁育和保护提供更精确的科学依据[22-24]。近些年,国内缅甸蟒野外数量急剧减少,而对该种原有分布区遗传学、分子生物学等相关研究十分缺乏。本研究对缅甸蟒养殖种群的遗传学参数等进行研究,为人工繁育的缅甸蟒种群健康发展和野生资源的迁地保护提供参考。
1 材料与方法
1.1样品采集与DNA提取
本研究样本来自海南东盛弘蟒业科技股份有限公司文昌养殖基地,采用无损伤取样法收集缅甸蟒蛇蜕样本,共42份,来自42个不同亲本。利用经典的苯酚-氯仿的方法提取缅甸蟒的蛇蜕基因组DNA[25],用分光光度计确定DNA的浓度,用灭菌水将DNA稀释至50 ng/μL,-20℃保存备用。
1.2微卫星引物筛选及扩增
参照Groot和Collins等共合成17对引物(PM-1、PM-2、PM-3、PM-5、30、Nsμ-3、Nsμ-10、MS5、MS6、MS9、MS10、MS11、MS13、MS16、MS19、MS22、MS24)[15-16],用于扩增缅甸蟒的基因组DNA,其中11对能够稳定扩增,但仅有8对具有多态性,选用这8对位点用于本次研究(表1)。上游或者下游引物的5′端加上FAM荧光(M13,5′-TGT AAA ACG ACG GCC AGT-3′)标记。所有引物均在上海生工生物公司合成。
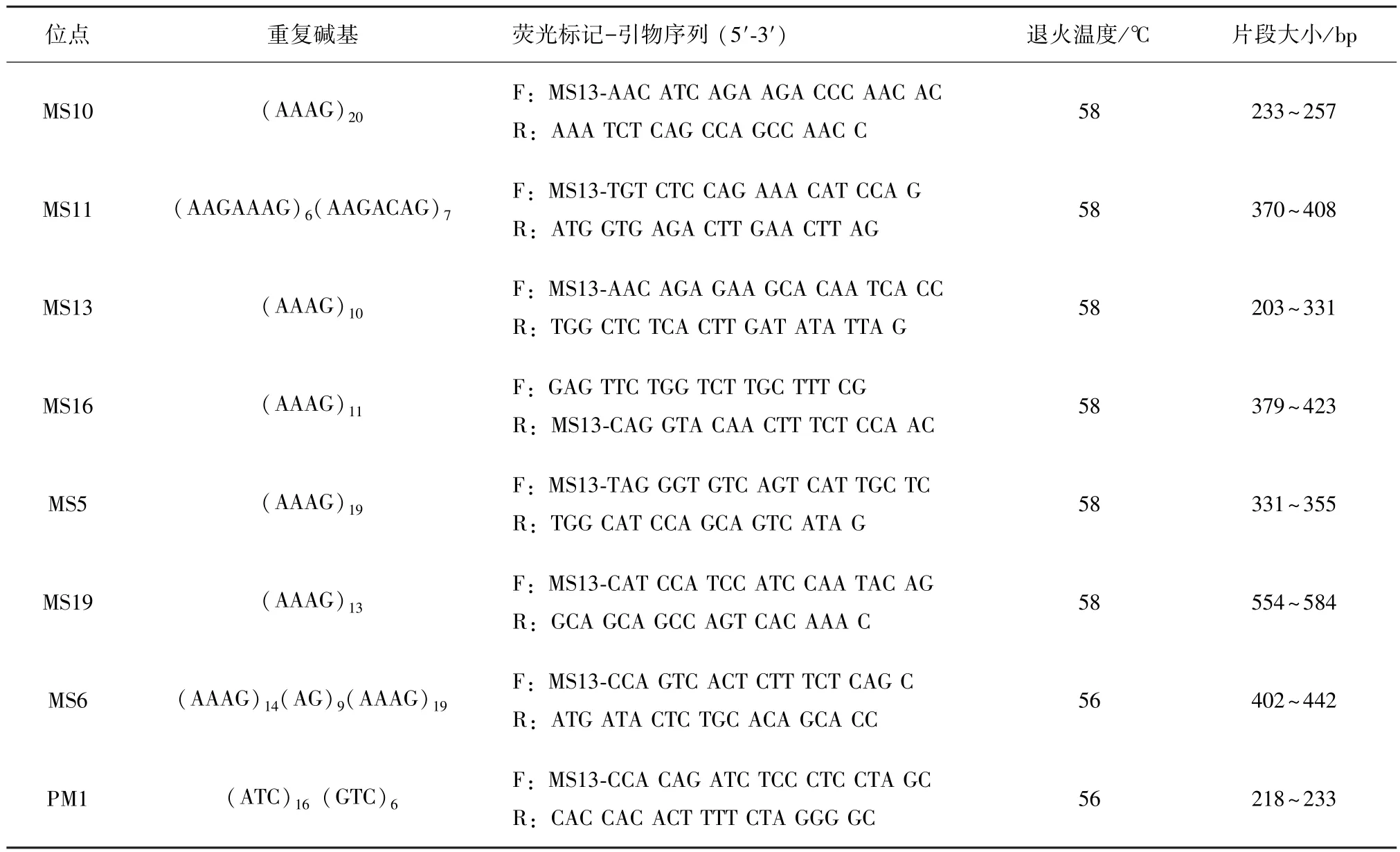
表1 缅甸蟒8对微卫星引物的信息(n=42)Table 1 Information of 8 pairs of microsatellite primers of Python bivittatus(n=42)
PCR反映采用12.5μL体系,内含1.25μL 10× buffer(Mg2+plus),1.25 μL dNTP(2 mmol/ L),0.2 μL F-Primer(10 μmol/ L),0.2 μL R-Primer (10 μmol/ L),0.1μL MS13-Tag(10μmol/ L),0.1μL EasyTaq DNA聚合酶(5 U/μL),1 μL DNA模板(50 ng/μL),补充无菌水8.4 μL。PCR扩增均在Eppendorf梯度PCR仪上完成,按照以下参数进行:95℃预变性5 min;94℃变性30 s,56~58℃(表1)复性45 s,72℃延伸40 s,扩增36个循环;最后再72℃延伸8 min,4℃保温。取反应产物2 μL,在ABI3730xl遗传自动分析仪(Applied Biosystems,美国)上对微卫星扩增产物进行分析。电泳结果用GeneMapper 4.0读取。
1.3数据统计
根据分子量数据确定个体各位点基因型,用POPGEN3.2计算等位基因数(Na),有效等位基因数(Ne),观测杂合度(Ho),期望杂合度(He)[26];用CERVUS 3.0计算多态信息含量(PIC),并采用马尔科夫链方法进行Hardy-Weinberg平衡检验[27]。
根据各位点等位基因频率,用BOTTLENECK 3.4进行瓶颈效应分析,基于无限等位基因模型(IAM)、双相突变模型(TPM)及逐步突变模型(SMM)计算平均期望杂合度(Heq),重复1 000次,采用符号检验和Wilcoxon符号秩次检验分析杂合过度是否显著,通过分析群体突变-漂移平衡来估计群体数量动态变化[28-29]。使用Populations 1.2.32(http:/ / bioinformatics.org/~tryphon/ populations/)和Treeview计算个体间遗传距离,根据遗传距离生成NJ聚类图[30]。
2 结果与分析
2.1微卫星引物在缅甸蟒中的PCR扩增结果
所选的17对微卫星引物中11对在中国缅甸蟒的养殖(HR)种群中获得了扩增产物。除了PM2、PM3、30表现单态外,其余8对微卫星引物在所有群体中均表现不同程度的多态性,根据各位点荧光引物PCR扩增产物ABI3100 Genetic Anslyzer检测结果,最终选择8个位点的遗传信息进行详细分析(表1)。
2.2微卫星遗传多样性
海南笼养种群的遗传多样性参数见表2。8个微卫星座位共检测出76个等位基因,平均等位基因数9.5个。其中,每对引物检测到的等位基因5~12个不等,有效等位基因数5.78。观测杂合度范围0.595~0.929,期望杂合度范围为0.734~0.851。
多态信息含量指标(PIC)值介于0.678和0.826之间,根据Botstein等提出的衡量基因变异程度高低的PIC[31],所选取的8个位点均为高度多态位点,说明可以有效进行遗传多样性分析。
2.3Hardy-Weinberg平衡
8个微卫星位点在缅甸蟒种群进行Hardy-Weinberg平衡检测(表2),位点MS10、MS13、MS6不同程度地偏离Hardy-Weinberg平衡(p<0.05),其中位点MS10、MS13处于极显著不平衡状态p<0.01)。在这3个位点中,MS10是由于杂合子过剩引起的,而MS13和MS6是由于杂合子缺失引起的。群体内固定系数为Fis=0.028。
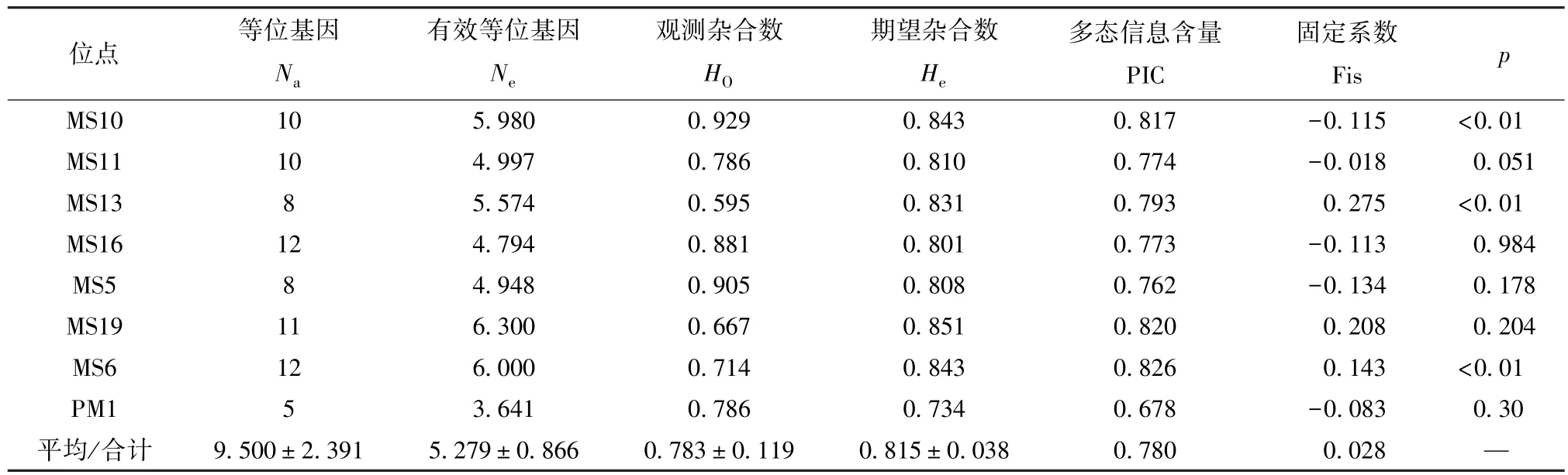
表2 缅甸蟒8对微卫星引物遗传多样性分析Table 2 Genetic diversity comparison of 8 pairs of microsatellite primers of Python bivittatus
2.4瓶颈效应分析
缅甸蟒养殖群体标记检验和标记秩检验结果,在IAM、TPM和SMM 3种突变模型假设下的平均期望杂合度(Heq)如表3所示,8个位点中,在IAM假设下,位点MS10、MS11、MS13、MS5、MS19、PM1的He均高于Heq,所有位点的He和Heq差异不显著(p > 0.05);在TPM假设下,位点MS10、MS13、MS5、PM1的He均高于Heq,且在MS16位点差异显著(p<0.05);在SMM假设下,位点MS13、PM1的He值高于Heq,其余的位点He低于Heq,且在MS16位点差异极显著(p<0.01),MS11、MS6位点差异显著(p<0.05)。Sign Test和Wilcoxon Test的结果如表4所示,虽然出现了杂合过剩的位点,但在IAM、TPM、SMM模型假设条件下,Sign Test和Wilcoxon Test均显示缅甸蟒养殖群体未显著偏离突变-飘移平衡(p > 0.05)。
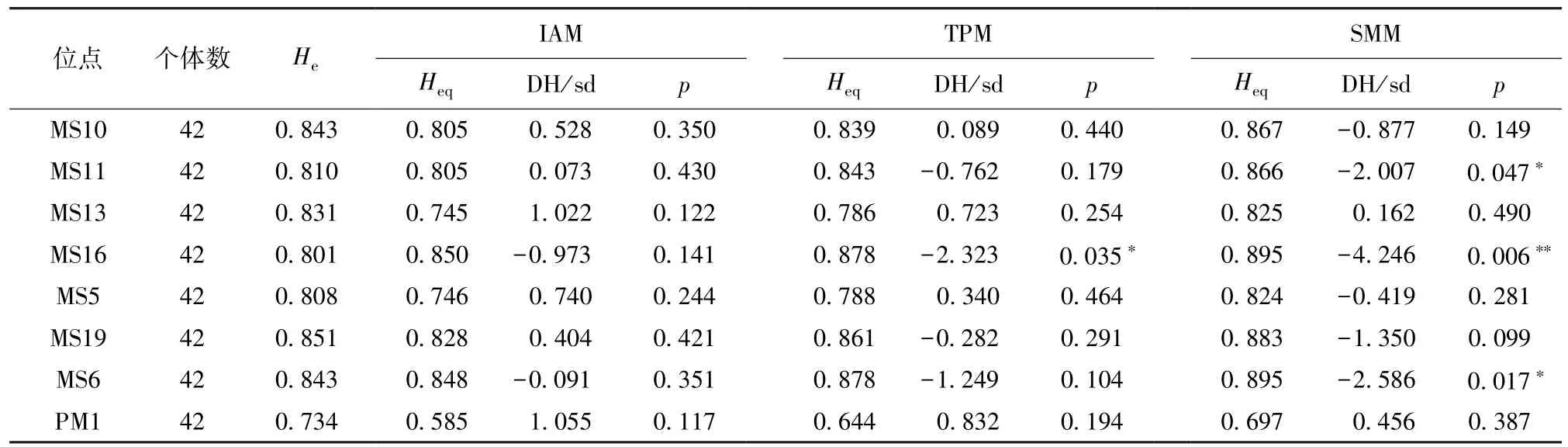
表3 缅甸蟒种群微卫星瓶颈效应分析Table 3 Microsatellite analysis of bottleneck effect in breeding populations of Python bivittatus

表4 缅甸蟒养殖群体突变-漂移平衡分析Table 4 Analysis on the test of departure from mutation-drift equilibrium in breeding populations of Python bivittatus
2.5个体遗传距离和聚类分析
缅甸蟒个体NJ聚类见图1。
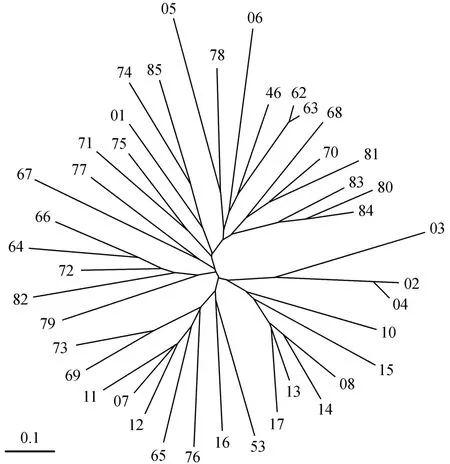
图1 缅甸蟒养殖个体NJ聚类Fig.1 NJ cluster in breeding individual of Python bivittatus
缅甸蟒个体间遗传距离变化范围为0.062 5~1,平均值0.66,其中遗传距离小于0.5占7.78%,0.5~0.6占16.38%,0.6~0.7占27.64%,0.7~0.8占24.27%,大于0.8占23.93%。从图1可见所有个体分布在3个不同的分支上,其中1个分支较大有24个个体,另外2个分支均由9个个体组成。
3 结论与讨论
对于野生动物的迁地保护来说,为了保持其遗传性状防止退化而进行遗传多样性的研究是必要的,其中杂合度是反映遗传多样性高低的重要指标之一[32]。根据研究结果,该养殖种群He值为0.815,高于大沼泽国家公园Collins研究的He(0.634)、Hunter研究的He(0.25~0.80)和越南的He(0.78)[15,17]。此外,与我国同一保护级别的其他物种遗传多样性进行比较,缅甸蟒He值也高于海南坡鹿(0.042~0.640)、大熊猫(0.483~0.704)、黄腹角雉(0.54)、扬子鳄(0.551)[33-36]。比近缘物种牙买加虹蚺(Epicrates subflavus)的He(0.57~0.64)略高,与马达加斯加树蚺(Sanzinia madagascariensis madagascariensis)He(0.825)、环颈蛇(Diadophis punctatus)He(0.816~0.962)相差不大[37-39]。可见海南人工养殖的缅甸蟒种群的遗传多样性处于较高的水平。在Hardy-Weinberg平衡检验中,大部分位点处于平衡状态,可能由于该养殖基地种蟒繁殖力较强,在选育过程中并未受到数量、选择压力的限制,此外在种蟒交配繁殖过程中,注重分子遗传学的应用,有效地避免了近交等因素的影响。
杂合子过剩是检验瓶颈效应的有效方法之一[29]。而微卫星数据已被证明更符合TPM模型用于群体数量瓶颈效应分析[40],本研究仅有位点MS16的He低于Heq且差异显著。从符号检验和Wilcoxon符号秩次检验的结果来看,在IAM和TPM 2种进化模式下检验表明,该养殖群体均没有显著的杂合过剩位点。本研究结果表明,缅甸蟒海南养殖群体在近期没有经历过瓶颈效应,群体数量没有出现过明显下降。
杂交优势是动物繁育过程中一种普遍现象,杂交优势大小一定程度上取决于双亲的遗传距离[41]。本研究个体间的遗传距离主要分布在0.5~1(92.22%),表明在以往的繁育过程中近交概率较小。在以后的种群选育过程中可选取距离较远的个体进行配对繁殖[42],同时,建立谱系管理制度,有效避免近交衰退,保持较丰富的遗传多样性。
[参考文献]
[1]赵尔宓.中国蛇类[M].合肥:安徽科学技术出版社,2006.
[2]IUCN.IUCN Red List of Threatened Species.Version 2014.2[EB/ OL].[2014-10-23].http:/ / www.iucnredlist.org.
[3]David G B,Tracy M B.The distribution of the Burmese python,Python molurus bivittatus[J].The Bulletin of the Chicago Herpetological Society,2008,43:33-38.
[4]David G B,Tracy M B.The distribution of the Burmese python,Python bivittatus[J].The Bulletin of the Chicago Herpetological Society,2010,45:86-88.
[5]国家林业局.中国重点陆生野生动物资源调查[M].北京:中国林业出版社,2009.
[6]Astarita G,Rourke B C,Andersen J B,et al.Postprandial increase of oleoylethanolamide mobilization in small intestine of the Burmese python(Python molurus)[J].American Journal of Physiology-Regulatory,Integrative and Comparative Physiology,2006,290:1407-1412.
[7]Mauldin R,Savarie P J.Acetaminophen as an oral toxicant for Nile monitor lizards(Varanus niloticus)and Burmese pythons(Python molurus bivittatus)[J].Wildlife Research,2010,37:215-222.
[8]Mccue M D,Bennett A F,Hicks J W.The effect of meal composition on specific dynamic action in Burmese Pythons(Python molurus)[J].Physiological and Biochemical Zoology,2005,78:182-192.
[9]Wang T,Altimiras J,Axelsson M.Intracardiac flow separation in an in situperfused heart from Burmese python(Python molurus)[J].The Journal of Experimental Biology,2002,205:2715-2723.
[10]Ratnoff O D,Rosenberg M J,Everson B,et al.Notes on clotting in a Burmese python(Python molurus bivittatus)[J].The Journal of laboratory and clinical medicine,1990,115:629-635.
[11]Coatoe T A,deKoning A P,Hall K T,et al.Sequencing the genome of the Burmese python(Python molurus bivittatus)as a model for studying extreme adaptations in snakes[J].Genome Biology,2011,12:406.
[12]Coatoe T A,Streicher J W,Meik J M,et al.Thousands of microsatellite loci from the venomous coralsnake Micrurus fulvius and variability of select loci across populations and related species[J].Mol Ecol Resour,2012,12:1105-1113.
[13]Castoe T A,Poole A W,deKoning A P,et al.Rapid microsatellite identification from illumina paired-end genomic sequencing in two birds and a snake[J].PLoS ONE,2012,7:e30953.
[14]Hunter M E,Hart K M.Rapid microsatellite marker development using next generation pyrosequencing to inform invasive Burmese python—Python molurus bivittatus—Management[J].International Journal of Molecular Sciences,2013,14:4793-4804.
[15]Groot T V,Bruins E,Breeuwer J A.Molecular genetic evidence for parthenogenesis in The Burmese python,Python molurus bivittatus[J].Heredity,2003,90:130-135.
[16]Collins T M,Freeman B.Genetic characterization of populations of the nonindigenous Burmese python in Everglades National Park[G].Miami:Florida International University,2008.
[17]Jordan P W,Goodman A E,Donnellan S.Microsatellite primers for Australian and New Guinean pythons isolated with an efficient marker development method for related species[J].Molecular Ecology Notes,2002,2:78-82.
[18]王志方,曹承和,李洁,等.蟒蛇的2个Dmrt基因的克隆及序列分析[C].中国动物学会两栖爬行动物学分会2005年学术研讨会暨会员代表大会论文集,2005.
[19]You C W,Lin Y P,Lai Y H,et al.Return of the pythons:first formal records,with a special note on recovery of the Burmese python in the demilitarized Kinmen islands[J].Zoological Studies,2013,52(1):1-11.
[20]柯文灿,武蕾蕾,吴文珊,等.基于Cytb和COI基因序列的蟒蛇分子鉴定[J].福建师范大学学报(自然科学版),2014,30(1):91-97.
[21]刘雅芳,曹婷,刘建才,等.缅甸蟒线粒体基因组全序列测定与分析[J].四川动物,2014,33(1):23-30.
[22]Gibbs H L,Prior K A,Weatherhead P J,et al.Genetic structure of populations of the threatened eastern massasuaga rattlesnake,Sistrurus catenatus:evidence from microsatellite DNA markers[J].Molecular Ecology,1997,6:1123-1132.
[23]Paquin M M,Wylie G D,Routman E J.Population structure of the giant garter snake,Thamnophis gigas [J].Conservation Genetics,2006,7:25-36.
[24]Marshall J C,Kingsbury B A,Minchella D J.Microsatellite variation,population structure,and bottlenecks in the threatened copperbelly water snake[J].Conserv Genet,2009,10:465-476.
[25]Saghai-Maroof M,Soliman K,Jorgensen R A,et al.Ribosomal DNA spacer-length polymorphisms in barley:Mendelian inheritance,chromosomal location,and population dynamics[J].Proceedings of the National Academy of Sciences,1984,81(24):8014-8018.
[26]Nei M.Estimation of average heterozygosity and genetic distance from a small number of individuals[J].Genetics,1978,89:583-590.
[27]Kalinowski T,Taper M L,Marshall T C.Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment[J].Mol Ecol,2007,16:1099-1106.
[28]Maruyama T,Fuerst P A.Population bottlenecks and nonequilibrium models in population genetics.II.Number of alleles in a small population that was formed by a recent bottleneck[J].Genetics,1985,111:675-689.
[29]苏翔,汪桂玲,冯建彬,等.鄱阳湖日本沼虾15个群体遗传多样性的微卫星分析[J].生态学杂志,2011,30(9):2007-2013.
[30]Takezaki N,Nei M.Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA[J].Genetics,1996,144:389-399.
[31]Botstein D,White R L,Skalnick M H,et al.Construction of a genetic linkage map in man using restriction fragment length polymorphism[J].American Journal of Human Genetics,1980,32:314-331.
[32]Cornuet J M,Luikart G.Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data[J].Genetics,1996,144:2001-2014.
[33]Dong L,Niu W,Zhou Z,et al.Assessing the genetic integrity of captive and wild populations for reintroduction programs:the case of Cabot’s Tragopan in China [J].Chinese Birds,2011,2(2):65-71.
[34]王芳,彭真信,张金国,等.应用微卫星标记分析圈养大熊猫遗传多样性[J].生物化学与生物物理进展,2007,34(12):1279-1287.
[35]张琼,吉亚杰,曾治高,等.奠基者效应对海南坡鹿迁地保护种群遗传多样性的影响[J].动物学杂志,2007,42(3):54-60.
[36]王义权,朱伟铨,王朝林.扬子鳄饲养种群线粒体DNA控制区的序列多态性[J].遗传学报,2003,30(5):425-430.
[37]Tzika A C,Koenig S,Miller R,et al.Population structure of an endemic vulnerable species,the Jamaican boa(Epicrates subflavus)[J].Molecular Ecology,2008,17:533-544.
[38]Ramanana M A,Bailey C A,Shore G D,et al.Characterization of 20 microsatellite marker loci in the Malagasy tree boa(Sanzinia madagascariensis madagascariensis)[J].Conservation Genetics,2009,10(6):1953-1956.
[39]Fontanella F M,Siddall M E.Isolation and characterization of 14 polymorphic microsatellite loci in the ringneck snake Diadophis punctatus(Colubridae:Dipsadinae)[J].Conserv Genet,2010,11:1193-1195.
[40]Di Rienzo A,Peterson A C,Garza J C.Mutational processes of simple sequence repeat loci in human populations[J].Proceedings of the National Academy of Sciences of the USA,1994,91:3166-3170.
[41]钱惠荣,康乐.DNA标记和分子育种[J].生物工程进展,1998,18(3):12-18.
[42]谷晶晶,朱迎军,孟雪松,等.微卫星标记对红鳍东方鲀繁殖的指导应用及遗传分析[J].水产科学,2010,29(9):527-531.
(责任编辑张坤)
第1作者:段玉宝(1984—),男,博士,讲师。研究方向:保护生物学和动物生态学。Email:ybduan@swfu.edu.cn。
Analysis on Genetic Diversity in Breeding Populations of Python bivittatus
Duan Yubao1,2,Wang Yingshu3,Ma Jianzhang2,Bai Suying2,Rong Ke2
(1.Key Laboratory of Forest Disaster Warning and Control of Yunnan Province,College of Forestry,Southweat Forestry University,Kunming Yunnan 650224,China;2.College of Wildlife Resource,Northeast Forestry University,Harbin Heilongjiang 150040,China;3.Shanghai Wild Animal Park,Shanghai 201399,China)
Abstract:Genetic diversity of breeding populations of Burmese python(Python bivittatus)were investigated using microsatellite DNA loci.The study showed that 76 alleles were detected among 42 sampled individuals,the average number of effective allele was 5.78,average of expected heterozygosity was 0.815,and average of PIC was 0.78.It was indicated that all the 8 loci were highly polymorphic,and only 3 loci were obviously departured from Hardy-Weinberg equilibrium High level of genetic diversities was represented in breeding popution of Python bivittatus)in Hainan.Mutation-drift equilibrium tests indicated that loci with significantly heterozygote excesses in populations,and the population was not obviously departured from mutation-drift equilibrium.It might indicate that the population did not suffer bottleneck effects in recent time,and no significant decline in the number of populations.
Key word:Python bivittatus,microsatellite,genetic diversity,breeding populations
通信作者:戎可(1972—),男,博士,副教授。研究方向:行为生态学和种群生态学。Email:rongke@nefu.edu.cn。
基金项目:国家自然科学基金项目(31372209、L1322010)资助;海南省科技厅产学研项目(CXY20130027)资助;西南林业大学博士科研启动金资助;西南林业大学云南省省级重点学科(林学)资助;西南地区生物多样性保育国家林业局重点实验室开放基金资助。
收稿日期:2015-12-28
doi:10.11929/ j.issn.2095-1914.2016.03.028
中图分类号:S718.61
文献标志码:A
文章编号:2095-1914(2016)03-0163-06
