COL1A2新突变致胎儿严重成骨不全症1例报告并表型基因型关联性文献复习
2016-05-09辰刘姜睿璇杨琳任芸芸周文浩
董 辰刘姜睿璇杨 琳任芸芸周文浩
COL1A2新突变致胎儿严重成骨不全症1例报告并表型基因型关联性文献复习
董辰1,4刘姜睿璇1,4杨琳1任芸芸2周文浩3
摘要目的总结1例COL1A2新突变致胎儿严重成骨不全症(OI)的临床特征及基因突变的特点,为胎儿产前咨询提供依据。方法对产检B超检查示OI可能的胎儿流产组织抽提DNA进行基因型分析,自行设计COL1A1和COL1A2所有外显子及剪接区域的引物。利用Sanger测序法对胎儿行COL1A1和COL1A2基因外显子及剪接区域的测序分析并行父母验证。依据人类基因突变数据库(HGMD)专业版,对COL1A2突变所致疾病临床表型行文献复习。结果胎儿的COL1A1和COL1A2基因均检测出变异位点。COL1A2基因检测到杂合突变(c.3142G>T,p.Glu1048Cys)在寡核苷酸多态性数据库、HGMD及Ⅰ型胶原蛋白突变数据库均未见报道,结合胎儿父母验证为新发突变,对比公共数据库及在线预测软件预测该突变类型为致病突变。在HGMD专业版中搜索COL1A2,共检索到387个COL1A2致病突变,与21种疾病及其亚型相关。92%的突变引起OI或其亚型,还可引起Ehlers-Danlos综合征或其亚型。结合COL1A2突变所致疾病临床表型行文献复习,本文报告胎儿符合Ⅱ型OI。结论产前通过超声影像结合基因分型诊断胎儿为COL1A2基因新发突变(c.3142G>T,p.Glu1048Cys)所致Ⅱ型OI; COL1A2基因编码蛋白长链双螺旋的400~480氨基酸及MLBR 3区域中甘氨酸被天冬氨酸或谷氨酸替代,多导致严重表型的OI;本文为产前准确预测胎儿结局、指导临床决策提供依据。
关键词COL1A2基因;成骨不全症;突变;表型基因型相关性分析
作者单位1复旦大学附属儿科医院上海,201102; 2复旦大学附属妇产科医院上海,200011; 3上海市出生缺陷防治重点实验室,复旦大学儿童发育与疾病转化医学研究中心,复旦大学附属儿科医院上海,201102; 4共同第一作者
1 病例资料
孕妇,G2P0,34岁。第1胎为自然胎停,未行相应检查。胎儿父亲39岁,体健。父母家族否认疾病家族史及近亲结婚史。孕妇孕早期未见异常,无放射性物质接触史,无药物服用史。孕24+1周于复旦大学附属妇产科医院行产检B超示:单胎,胎儿头位,可见胎心胎动;胎头双顶径长58 mm,头围204 mm,腹围173 mm;右侧肱骨长32 mm,左侧肱骨长29 mm;右侧脑室后角3.9 mm;小脑横径25 mm;后颅窝池深5.9 mm;可见局部肠管强回声;左侧股骨长21 mm,见弯曲;可见右侧股骨长30 mm,明显弯曲、成角畸形(图1A)。依据单胎、局部肠管强回声、股骨及肱骨短小、双侧股骨成角弯曲,产检B超提示成骨不全症(OI)可能。
经胎儿产前诊断与咨询,孕妇选择行人工流产术终止妊娠,并同意留取胎儿流产组织行分子诊断,先在上海集爱遗传与不育诊疗中心行人类全基因组SNP分型芯片检测,未见明显染色体拷贝数异常。为求得进一步诊断来复旦大学附属儿科医院(我院)行基因检测。经胎儿家属同意及我院伦理委员会审核通过,取胎儿流产组织,父、母亲外周静脉血各2mL(EDTA抗凝),抽提DNA(TIANGEN试剂盒抽提胎儿组织DNA,QIAGEN mini blood全血试剂盒抽提父母外周血DNA),行基因分析。基于文献复习,约90%的OI是由常染色体显性遗传COL1A1和COL1A2基因突变引起[1],自行设计COL1A1 (NM_000088)和COL1A2 (NM_ 000089)所有外显子及剪接区域的引物72(35和37)对,全部引物由上海捷瑞生物工程有限公司合成。利用Sanger测序法(3500XL Genetic Analyzer,ABI),对该胎儿分别行COL1A1和COL1A2基因外显子及剪接区域的测序分析。
胎儿2个基因上均检测出变异位点,对照寡核苷酸多态性数据库(dbSNP)、人类基因突变数据库(HGMD)和Ⅰ型胶原蛋白突变数据库(http: / /www.le.ac.uk/genetics/ collagen/)确定变异位点性质。图1B~F显示患儿及其父母COL1A2突变位点在染色体及蛋白质位置,胎儿COL1A1基因中有5个SNP位点和1个剪接区域的未知突变: c.1156-9G>A,COL1A2基因中有6个SNP位点和1个编码区致病性未知的突变: c.3142G>T p.Glu1048Cys。COL1A2基因检测到的杂合突变(c.3142G>T,p.Glu1048Cys)在dbSNP、HGMD及Ⅰ型胶原蛋白突变数据库均未见报道。胎儿父母验证此位点无突变,证实为新发突变(图1F)。对比公共数据库,ExAC及1000 Genomes phase数据库中均未有该突变,并且在线预测软件Mutation Taster(http: / /www.mutationtaster.org/)预测该突变类型为致病突变。

图1 胎儿B超图像和胎儿及其父母COL1A2突变位点在染色体及蛋白质位置分析注 A:胎儿B超图像,所示右侧股骨成角畸形; B: 7号染色体结构示意图; C: COL1A2基因所编码蛋白质全长及其功能结构域:绿色代表胶原三螺旋重复区域,紫红色代表胶原C端结构域; D: HGMD报道的COL1A2基因编码区错义突变导致不同疾病的突变位点位置示意图:红色条柱代表导致严重表型的Ⅱ型和Ⅲ型OI的突变位点,绿色条柱代表导致表型较轻的Ⅰ型和Ⅳ型OI的突变位点,黄色条柱代表除OI外导致其他疾病的突变位点; E:Ⅰ型胶原蛋白的结构示意图:灰色代表两条由COL1A1编码的α1肽链,红色代表由COL1A2编码的α2肽链; F:胎儿及其家系突变位点Sanger验证峰图,证实该位点为新发(de novo)。图B~F中的红色柱分别代表该突变位点在染色体、α1肽链、HGMD报道的突变位点、Ⅰ型胶原和Sanger测序碱基序列中的位置
2 数据库整理
截至2016年1月20日,在HGMD专业版中搜索COL1A2,共检索到387个HGMD收录的COL1A2基因致病突变,与21种疾病及其亚型相关。共包含279个编码区错义突变,11个编码区小片段缺失突变,11个编码区小片段插入突变,5个编码区相邻位置双突变,81个非编码区突变。
结合既往文献报道及在线人类孟德尔遗传数据库(OMIM),表1总结了HGMD中报道的COL1A2突变所引起的主要疾病种类及临床表型。92%的突变引起OI或其亚型,6%的突变引起Ehlers-Danlos综合征(EDS)或其亚型,1例引起Marfan综合征(c.2123G>A,p.Arg708Gln)[2],1例引起青年型骨质疏松(c.1576G>A,p.Gly526Arg)[3],1例引起骨质疏松症(c.2251G>A,p.Gly751Ser)[4]。除此之外,还有2例非编码区突变导致转录增加[5],1例与颅内动脉瘤易感性相关[6],1例导致轻微结缔组织异常[7]。
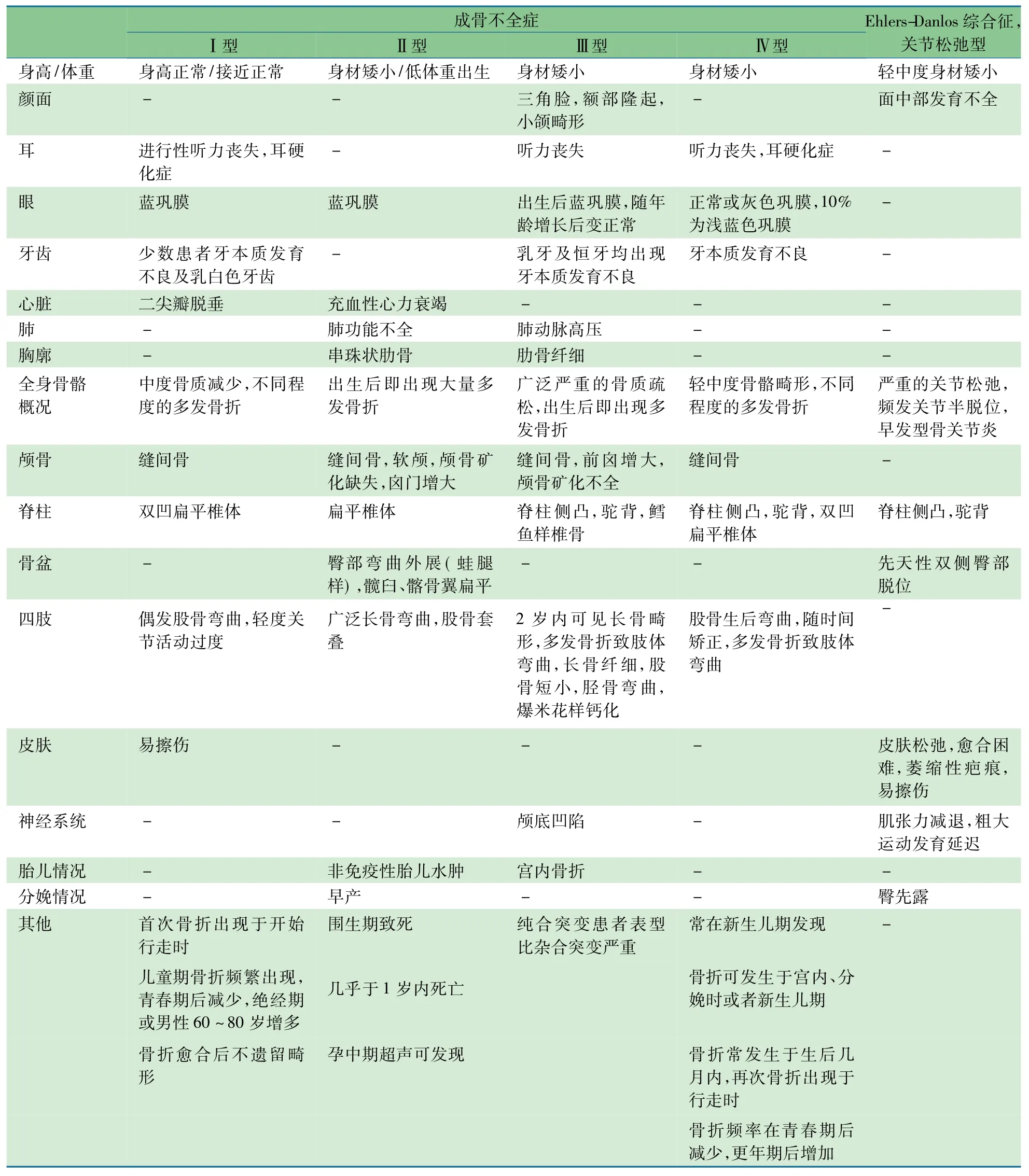
表1 COL1A2突变所致疾病临床表型比较
虽然目前的研究已将OI的分型增加到17种,但是COL1A2基因突变可引起Ⅰ、Ⅱ、Ⅲ、Ⅳ4种OI亚型,且引起的各型数量较为平均。其中76%是由错义突变引起,占所有错义突变引起疾病的98%。约20%的COL1A2基因的非错义突变(移码突变、整码突变和非编码区碱基突变)会导致EDS或其亚型,1例错义突变引起EDS(c.1201G>T,p.Glu1201Term)[8]。EDS根据产生的原因分为主要的6大类以及其他类型[9]。其中,COL1A2基因突变引起的主要是关节松弛型EDS。HGMD有1例为未分型的EDS[10],其余均为关节松弛型EDS。
本文胎儿超声影像初步诊断为OI的患儿进行分子诊断,结合既往文献、数据库及相关软件,诊断为COL1A2基因突变(c.3142G>T,p)所致的Ⅱ型OI。
3 讨论
COL1A2基因位于第7号染色体7q22.1,由52个外显子组成,编码骨骼中的Ⅰ型胶原的α2肽链。1条α2肽链与2条COL1A1基因编码的α1肽链组成异源三聚体,3条肽链相互缠绕成绳索样结构,中间的螺旋状功能域及两侧N端和C端的球状前肽链构成了Ⅰ型前胶原分子。螺旋状功能域是一个由1 014个氨基酸组成的Gly-Xaa-Yaa三肽重复的三重螺旋结构。只有最小的甘氨酸分子才能进入三重螺旋的内部结构。通过对肽链特定部位的羟化、折叠,以及分泌后对N端和C端前肽剪除的成熟过程,最终形成成熟Ⅰ型胶原。Ⅰ型胶原是构成骨骼、皮肤和肌腱等结缔组织的最主要的细胞外基质蛋白,在生长发育中起着重要的作用。
在COL1A2基因突变引起的多种疾病中,最多见的为OI。OI又称脆性骨病或脆骨-蓝巩膜-耳聋综合征,是一种罕见的以骨脆性增加和胶原蛋白代谢紊乱为特征的风湿性疾病[11]。1979年,Sillence等根据临床症状、影像学表现及分子生物学信息将OI为4型:Ⅰ型最主要的特点为蓝巩膜,Ⅳ型巩膜表现为正常,Ⅱ型为围生期致死型,Ⅲ型为进展型不伴有巩膜异常[12]。
表1总结的4种OI分型的临床表型中,①从骨骼表型看,Ⅰ和Ⅳ型OI的骨骼畸形程度明显小于Ⅱ和Ⅲ型,颅骨也仅出现缝间骨的改变,而没有囟门增大或者矿化异常。特别是Ⅰ型OI患儿并未出现身材矮小,四肢长骨的弯曲偶尔出现,骨折发生的时间较晚,频率也会随着性成熟而减少,愈合后也不会遗留畸形。虽然Ⅳ型OI的骨折发生时间较早,但发生的频率也会随着青春期之后而减少,更年期之后再增加。Ⅱ型OI患儿多存活不超过1岁,Ⅲ型OI患儿生后2年内即可发现骨骼畸形,广泛严重的骨质疏松使其出生后即出现多发骨折,骨折愈合后常遗留肢体畸形。②从生长发育来看,Ⅳ型OI的身材矮小主要表现为比正常人群的平均身高低5%,Ⅲ型OI成年人的身高在92~108 cm,远低于Ⅳ型OI患者。③从不同系统来看,Ⅱ、Ⅲ型常有呼吸、循环及神经系统的严重表型。总之,Ⅱ、Ⅲ型OI表型严重,Ⅰ、Ⅳ型OI表型轻微。
依据临床表型的不同,如果能早期通过分子诊断的方法明确疾病的分型,或可尽早干预或可提示预后。已有研究表明,COL1A1和COL1A2无义或者移码突变导致胶原数量减少而引起的OI,常为临床表型最轻微的Ⅰ型OI,而导致胶原结构异常的错义突变则更易导致致死型OI[13]。图1D总结了HGMD中COL1A2基因编码区所报道的所有错义突变,并根据突变位点及其导致的疾病进行表型基因型相关性分析。
在COL1A2基因C端的突变均导致表型轻微的Ⅰ、Ⅳ型OI或者其他综合征,而N端几乎没有突变会出现相关表型。而长链螺旋功能域的突变位点大多数都能导致疾病,而且表型轻重不一。研究还发现,这些位点的变异均是Gly-Xaa-Yaa三肽重复中甘氨酸被其他氨基酸替代。从胶原分子的结构中不难推断,甘氨酸残基是唯一可以进入螺旋内,调节三股螺旋核心空间结构的分子。因此,当甘氨酸被其他大分子氨基酸替代时,螺旋折叠结构就会被破坏,三条链就会暴露在外,过多的被修饰[14]。
进一步研究发现,除了甘氨酸被替代的变异,仅有2个错义突变会导致表型严重的Ⅱ型OI(分别是p.His182Asp 及p.Arg234Cys)[1,15],其余突变均导致的是轻型OI及其他综合征。在由丙氨酸或丝氨酸替代甘氨酸的突变中仅有少数为严重表型,而由半胱氨酸、缬氨酸和精氨酸替代的有近50%突变为严重表型,≥50%由天冬氨酸和谷氨酸替代的突变会表现出严重致死型的表型。可能与氨基酸的大小及其酸碱性相关,既往研究也表明相对分子质量相对较小的丙氨酸和丝氨酸对胶原结构的影响较小,而酸性氨基酸(天冬氨酸和谷氨酸)则影响较大。特别是那些在同一位点突变为不同氨基酸的突变,所导致的临床表型不同。如在氨基酸第286位Gly286Ala的患者表现为Ⅳ型OI[16],而Gly286Val的患者就表现为严重的Ⅱ型OI[13]。
另外在长链中出现轻重表型交叉,还有可能和转录后修饰的位置相关,如在氨基酸计数400~480区域较集中的出现所致严重表型的突变,就有可能是和第420位、441位及444位氨基酸转录后需要被羟化有关,在这一区域突变会使羟化过程受到干扰,从而导致严重表型。
本文患儿所检测到的突变位于c.3142G>T,p.Glu1048Cys,既往未报道。这一突变位于长链螺旋功能域,是Gly-Xaa-Yaa三肽重复中甘氨酸被半胱氨酸所替代。在氨基酸序列第1 048位前后目前已报道的突变中,均是导致严重表型的突变。本文胎儿在产检超声中已显示股骨弯曲畸形,属于Ⅱ型OI,符合严重临床表型,与既往文献报道相符。并结合基因突变在线预测软件Mutation Taster的结果,可确定该突变导致患儿Ⅱ型OI。查询既往文献该区域属于Ⅰ型胶原的主要配基结合区域3(MLBR 3)[17],该区域对胶原分子间交联、核心蛋白聚糖的结合都有重要作用[18,19],故推测该区域突变致严重表型的原因与蛋白相互作用的功能相关。
4 结论
本文通过产前超声影像结合基因分型诊断1例胎儿为COL1A2基因新发突变(c.3142G>T,p.Glu1048Cys)所致Ⅱ型OI。结合表型基因型相关性分析,可初步得出在COL1A2基因编码蛋白长链双螺旋的400~480氨基酸及MLBR 3区域中甘氨酸被天冬氨酸或谷氨酸替代,多会导致严重表型的OI。本文同时也为产前准确预测胎儿结局、指导临床决策提供依据。
参考文献
[1]Bodian DL,Chan TF,Poon A,et al.Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships.Hum Mol Genet,2009,18(3) : 463-471
[2]Phillips CL,Shrago-Howe AW,Pinnell SR,et al.A substitution at a non-glycine position in the triple-helical domain of pro alpha 2(I) collagen chains present in an individual with a variant of the Marfan syndrome.J Clin Invest,1990,86(5) : 1723-1728
[3]Dawson PA,Kelly TE,Marini JC.Extension of phenotype associated with structural mutations in type I collagen: siblings with juvenile osteoporosis have an alpha2(I) Gly436 -->Arg substitution.J Bone Miner Res,1999,14(3) : 449-455
[4]Spotila LD,Constantinou CD,Sereda L,et al.Mutation in a gene for type I procollagen (COL1A2) in a woman with postmenopausal osteoporosis: evidence for phenotypic and genotypic overlap with mild osteogenesis imperfecta.Proc Natl Acad Sci U S A,1991,88(12) : 5423-5427
[5]Akai J,Kimura A,Hata RI.Transcriptional regulation of the human type I collagen alpha2 (COL1A2) gene by the combination of two dinucleotide repeats.Gene,1999,239 (1) : 65-73
[6]Yoneyama T,Kasuya H,Onda H,et al.Collagen type I alpha2 (COL1A2) is the susceptible gene for intracranial aneurysms.Stroke,2004,35(2) : 443-448
[7]Oliver JE,Thompson EM,Pope FM,et al.Mutation in the carboxy-terminal propeptide of the Pro alpha 1(I) chain of type I collagen in a child with severe osteogenesis imperfecta (OI type III) : possible implications for protein folding.Hum Mutat,1996,7(4) : 318-326
[8]Schwarze U,Hata R,McKusick VA,et al.Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway.Am J Hum Genet,2004,74(5) : 917-930
[9]Beighton P,De Paepe A,Steinmann B,et al.Ehlers-Danlos syndromes: revised nosology,Villefranche,1997.Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK).Am J Med Genet,1998,77(1) : 31-37
[10]AAFP age charts for periodic health examinations: 40 to 64 years.Commission on Public Health and Scientific Affairs.Am Fam Physician,1992,45(4) : 1917-1920
[11]Byers PH.Osteogenesis imperfecta.In: Royce PM.; Steinmann B.Connective Tissue and Its Heritable Disorders: Molecular,Genetic,and Medical Aspects.New York: Wiley-Liss; 1993
[12]Sillence DO,Senn A,Danks DM.Genetic heterogeneity in osteogenesis imperfecta.J Med Genet,1979,16(2) : 101-116
[13]Marini JC,Forlino A,Cabral WA,et al.Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans.Hum Mutat,2007,28(3) : 209-221
[14]Engel J,Prockop DJ.The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper.Annu Rev Biophys Biophys Chem,1991,20: 137-152
[15]Galicka A,Woiczyński S,Gindzieński A.Studies on type I collagen in skin fibroblasts cultured from twins with lethal osteogenesis imperfecta.Acta Biochim Pol,2003,50(2) : 481-488
[16]Ward LM,Lalic L,Roughley PJ,et al.Thirty-three novel COL1A1 and COL1A2 mutations in patients with osteogenesis imperfecta types I-IV.Hum Mutat,2001,17(5) : 434
[17]Di Lullo GA,Sweeney SM,Korkko J,et al.Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human,type I collagen.J Biol Chem,2002,277(6) : 4223-4231
[18]Hanson DA,Eyre DR.Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone.J Biol Chem,1996,271(43) : 26508-26516
[19]Keene DR,San Antonio JD,Mayne R,et al.Decorin binds near the C terminus of type I collagen.J Biol Chem,2000 Jul 21; 275(29) : 21801-21804
(本文编辑:张崇凡)
A de novo COL1A2 gene mutation in a fetus with severe osteogenesis imperfect and phenotype-genotype correlation analysis
DONG Cheng1,4,LIUJIANG Rui-xuan1,4,YANG Lin1,REN Yun-yun2,ZHOU Wen-hao3(1 Children's Hospital of Fudan University,Shanghai 201102; 2 Obstertrics and Gynecology Hospital of Fudan University,Shanghai 200011; 3 The Molecular Genetic Diagnosis Center,Shanghai Key Lab of Birth Defect,Translational Medicine Research Center of Children Development and Disease,Pediatrics Research Institute,Children's Hospital of Fudan University,Shanghai 201102,China; 4 Co-first author)
Corresponding Author: ZHOU Wen-hao,E-mail: zwhchfu@126.com; REN Yun-yun,E-mail: renyunyun@ hotmail.com
AbstractObjective To summarize the clinical features and gene mutation characteristics of a de novo COL1A2 gene mutation in a fetus with severe osteogenesis imperfect (OI),and to provide evidence for prenatal counseling.Methods DNA was extracted from the fetal abortion tissues,and primers of the whole exons and splicing sites of COL1A1 and COL1A2 genes were designed.Using Sanger sequencing,the fetal sequences of the whole exons and splicing sites of COL1A1 and COL1A2 genes were analyzed and confirmed with the parents'samples.According to HGMD,literatures on clinical symptoms of the diseases caused by COL1A2 mutation were reviewed.Results The variants on both COL1A1 and COL1A2 gene were detected.On COL1A2 gene a de novo heterozygous mutation (c.3142G>T,p.Glu1048Cys) was detected,which had never been reported in dbSNP,HGMD and osteogenesis imperfecta&Ehlers-Danlos syndrome variant databases.Compared with the common database and online prediction software the mutation was predicted to be a the disease-causing mutation.HGMD professional version was searched with "COL1A2" and 387 disease-causing mutations were found to be related to 21 diseases or their subtypes.Ninety-two percent of the mutations caused OI or its subtypes; others caused Ehlers-Danlos syndrome or its subtype.Combined with the clinical symptoms of the disease caused by COL1A2 gene,the fetus was more consistent with the OI typeⅡ.Conclusion A fetus OI typeⅡcaused by a de novo mutation in COL1A2(c.3142G>T,p.Glu1048Cys) was diagnosed with prenatal ultrasound imaging and genotyping.Glycine in 400 to 480 amino acid and MLBR 3 region of the protein encoded by COL1A2 gene replaced by aspartate acid or glutamic will causebook=43,ebook=46sever OI.This article provides the basis for accurate prediction of fetal outcome and clinical decision-making.
Key wordsCOL1A2 gene; Osteogenesis imperfect; Mutation; Phenotype genotype correlation analysis
(收稿日期:2015-10-30修回日期: 2016-01-27)
通讯作者周文浩,E-mail: zwhchfu@126.com;任芸芸,E-mail: renyunyun@ hotmail.com
基金项目上海市科学技术委员会: 14411950402;新生儿严重神经系统畸形规范化诊治的多中心临床研究;上海市科学技术委员会: 15XD1500800;上海市优秀学术带头人新生儿常见出生缺陷疾病的分子遗传研究
DOI:10.3969/j.issn.1673-5501.2016.01.011
