硼掺杂金刚石薄膜电极上二氯酚的电化学阻抗谱研究
2016-04-08吕江维曲有鹏冯玉杰刘峻峰
吕江维, 曲有鹏, 冯玉杰, 刘峻峰
(1. 哈尔滨商业大学药学院, 哈尔滨 150076;
2. 哈尔滨工业大学生命科学与技术学院, 哈尔滨 150080;
3. 哈尔滨工业大学城市水资源与水环境国家重点实验室, 哈尔滨 150090)
硼掺杂金刚石薄膜电极上二氯酚的电化学阻抗谱研究
吕江维1, 曲有鹏2, 冯玉杰3, 刘峻峰3
(1. 哈尔滨商业大学药学院, 哈尔滨 150076;
2. 哈尔滨工业大学生命科学与技术学院, 哈尔滨 150080;
3. 哈尔滨工业大学城市水资源与水环境国家重点实验室, 哈尔滨 150090)
摘要以2,4-二氯酚(2,4-DCP)和2,6-二氯酚(2,6-DCP)为模型污染物, 采用循环伏安法和电化学阻抗谱研究了硼掺杂金刚石(BDD)电极上2种氯酚的电催化氧化过程. 结果表明, 2,4-DCP和2,6-DCP的氧化电位分别为1.55和1.62 V. 等效电路拟合结果表明, 当极化电位由开路电位提高至1.5 V时, 2种氯酚的电荷转移电阻均有明显下降, 反应控制步骤为扩散控制步骤. 与2,6-DCP相比, 2,4-DCP在BDD电极上更容易发生直接电化学氧化.
关键词硼掺杂金刚石; 循环伏安; 电化学阻抗谱; 二氯酚; 电催化
20世纪90年代, Carey等[1]将硼掺杂金刚石(BDD)薄膜电极引入到废水处理中, 为采用电催化氧化法处理有机污染废水的研究提供了新方向. 与传统用于废水处理的电催化电极相比, BDD薄膜电极电化学势窗较宽, 析氧电位高[2~4], 可使难降解有机物转化为可降解物质或将其分解成无毒的二氧化碳; 且电极材料化学稳定性好, 不易污染[3], 可以长期使用不需更换; 电极降解有机污染物的反应速度快, 电流效率高, 生成的中间产物少[5,6].
目前, 制备BDD薄膜电极的化学气相沉积技术已日趋成熟[7~9], 研究工作侧重于探索适宜的降解工艺条件, 对电极的电化学性质和电催化机理等也有初步的研究[10~14]. BDD薄膜电极本身的电化学性质会显著影响污染物在BDD薄膜电极上的降解效果, 这种性质与电极的化学结构和微观状态等有关. 但由于不同的化学气相沉积工艺制备的BDD薄膜电极的电化学性质差异较大, 对于不同BDD薄膜电极的物理、化学和电子特性与其电化学行为关系的研究仍然不够完善[15]. 明确BDD薄膜电极与溶液界面的结构和性质, 了解电极的电化学行为才能有效地控制电极反应的性质和速度. 电化学阻抗谱是研究电极过程及电极界面现象的有效方法. 赵国华等[16]采用电化学阻抗谱研究了不同电极电位下苯酚在BDD薄膜电极表面形成聚合物薄膜的过程, 发现不同电位下反应动力学过程的控制步骤不同; 另外还发现在铁氰化钾电解液中的电化学反应具有良好的准可逆性[17]. 刘峰斌等[18]对表面氢吸附和氧吸附的BDD薄膜电极的电化学阻抗谱进行了研究, 发现氧吸附BDD薄膜电极的空间电荷层电阻和电容更大, 极化电阻也比氢吸附金刚石薄膜电极要大. 崔凯等[19]采用交流阻抗法发现自组装纳米金颗粒修饰后的BDD薄膜电极阻抗明显减小.
本文采用电化学阻抗谱并结合循环伏安法, 以2种二氯酚为模型污染物, 分析了二氯酚在BDD薄膜电极/溶液界面上的电化学过程及相关的电极动力学参数, 对研究BDD薄膜电极降解污染物的机制具有参考价值.
1实验部分
1.1试剂与仪器
氢气和甲烷(纯度99.999%), 哈尔滨黎明气体集团; 2,4-二氯酚(2,4-DCP)和2,6-二氯酚(2,6-DCP)为分析纯, 美国Sigma公司; 硼酸三甲酯(分析纯), 上海阿拉丁生化科技股份有限公司; Na2SO4(分析纯), 汕头西陇化工厂; 无水乙醇, 哈尔滨新达化工厂. 溶液均由超纯水配制. 硅片, 上海君合电子材料有限公司. Ti板, 宝鸡宝冶钛镍制造公司.
Model 263A型电化学工作站(美国普林斯顿公司), 采用三电极体系进行电化学分析, 工作电极为BDD薄膜电极, 辅助电极为Pt电极(北京有研亿金公司), 参比电极为Ag/AgCl电极(电位为0.2224 V, 上海仪电科学仪器股份有限公司).
1.2实验过程
采用的基体材料为p型重掺杂硼的单晶硅片[电阻率6 mΩ·cm, 单面抛光处理, 晶向[111], 厚度(380±20) μm]. 沉积BDD薄膜前先对基体进行预处理: 首先用氢氟酸溶液对硅片表面进行漂洗, 以除去表面的自然氧化层; 经蒸馏水清洗后, 再用金刚石研磨膏对硅片进行研磨; 最后用乙醇溶液超声清洗10 min, 将表面残留的金刚石颗粒洗去, 晾干后待用.
BDD薄膜电极采用直流等离子体化学气相沉积法制备, 反应气体为高纯度的甲烷、氢气和硼酸三甲酯(体积比为4∶190∶10), 基片温度1000 ℃, 压强15.2~16.0 kPa, 电压720~730 V, 电流7.8~8.4 A, 沉积时间为11.5 h. 电极制备的实验装置见文献[20], 具体制备参数与文献[20]中的样品c相同. 将制备好的BDD薄膜背面用细砂纸打磨, 去除在薄膜生长过程形成的碳膜, 露出平整的Si表面, 然后用导电银胶将Si表面粘到Ti板上, 使Si表面和Ti板形成良好的接触.
2结果与讨论
2.1二氯酚的循环伏安特性
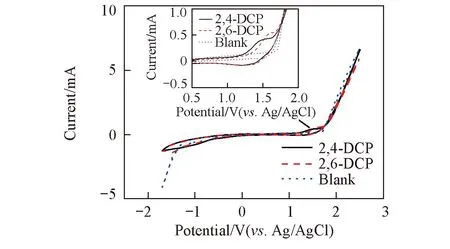
Fig.1 Cyclic voltammograms of BDD electrodes in 1 mmol/L 2,4-DCP+0.1 mol/L Na2SO4 and 1 mmol/L 2,6-DCP+0.1 mol/L Na2SO4 solutions
循环伏安测试的溶液分别为1 mmol/L 2,4-DCP+0.1 mol/L Na2SO4和1 mmol/L 2,6-DCP+0.1 mol/L Na2SO4, 扫描速度100 mV/s, 扫描电位范围-1.7~2.5 V. 空白实验为只含电解质Na2SO4不含2种二氯酚的溶液, 其它测试条件完全相同. 结果如图1所示.
从图1可以看出, 2种二氯酚在正向扫描时均出现了氧化峰, 2,4-DCP及2,6-DCP的氧化峰电位分别为1.55和1.62 V, 超过2.0 V后开始发生析氧反应; 在负向扫描时, 2,4-DCP和2,6-DCP出现了弱还原峰. 2种二氯酚的氧化峰和还原峰电流值均不相等, 并且峰电位差值均大于59 mV, 因此2种二氯酚在BDD电极上的反应均为不可逆反应.

Fig.2 Bode plots of BDD electrodes in 1 mmol/L 2,4-DCP+0.1 mol/L Na2SO4 solution at different potentials
2.2二氯酚的电化学阻抗谱
2.2.12,4-DCP的电化学阻抗谱电化学阻抗谱测试的溶液为1 mmol/L 2,4-DCP+0.1 mol/L Na2SO4, 测试正弦波频率范围为105~10-1Hz, 振幅为5 mV, 测试电位分别为开路电位和1.0, 1.5, 2.0和2.5 V.
由图1的CV测试结果可知, 开路电位是平衡电位, 没有发生氧化还原反应, 1.5 V左右是氯酚发生直接氧化反应的电势, 而1.0 V介于前两者之间, 2.0 V左右是发生析氧反应的电位, 而2.5 V是发生显著析氧反应的电位.
图2和图3是2,4-DCP在BDD薄膜电极上5个电位下的Bode图和Nyquist图. 从图2可以看出, 在开路电位、1.0 V和1.5 V电位下, 相位角对频率的曲线有2个峰, 推测至少有2个电容元件; 而2.0和2.5 V的曲线仅有1个峰, 推测至少有1个电容元件. 由图3中1.5 V的Nyquist曲线可以看出, 高频有1个容抗弧, 低频曲线为近似呈45°角的直线, 说明有扩散引起的韦伯(Warburg)阻抗. 图2和图3中高频端的阻抗是半导体BDD电极空间电荷层与溶液电阻的响应, 低频端的阻抗是电催化反应的响应. Nyquist图上低频阻抗的容抗弧对应一个状态变量, 即电极电位. 改变电极电位可以改变半导体表面的电子浓度. 随着阳极极化电位的增加, 低频容抗弧的直径明显减小, 即电催化反应的电阻减小, 这个容抗弧反映的是电催化速率的大小.
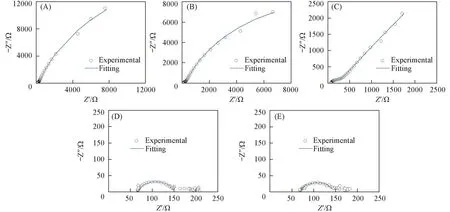
Fig.3 Nyquist plots of BDD electrodes in 1 mmol/L 2,4-DCP+0.1 mol/L Na2SO4 solution at different potentials (A) Open circuit potential; (B) 1.0 V; (C) 1.5 V; (D) 2.0 V; (E) 2.5 V.
电极反应主要发生在电极与溶液的界面上, 界面的结构和性质显著影响电极的电催化反应. 根据半导体的能带理论, 当半导体电极与电解质溶液接触后, 由于初始各自的费米能级不同, 电子在半导体和在电解质溶液中的氧化还原对中的能量不同, 电子将在半导体与溶液的界面之间发生转移, 从能量高的一侧转移到能量低的一侧, 在界面的两侧会形成剩余电荷, 从而建立起一个界面电场. 该界面电场将抑制电子进一步转移, 并最终达到平衡, 这时在半导体一侧形成了空间电荷层. 在溶液中, 由于两相中的剩余电荷有静电相互作用, 电极表面与溶液中的各种溶剂分子、溶剂化的离子和分子等粒子之间的短程作用, 溶液一侧也会形成电荷层, 这样的结构称为电极/溶液界面的双电层. 在半导体一侧剩余电荷的分布是分散的, 这主要是因为半导体中载流子浓度比较低. 当溶液中的电解质浓度也较低时, 溶液一侧的双电层也是分散的, 一般认为半导体/溶液界面由3种不同的电荷分布区组成: 半导体/溶液界面上的紧密双电层(Helmholtz层)、半导体一侧的空间电荷层及液相中的分散层[21]. BDD电极是p型半导体电极, 且电解质Na2SO4的浓度(0.1 mol/L)较低, 因此半导体/溶液界面两侧双电层的分布都是分散的. 电极/溶液界面的结构如图4所示. 通常在不施加电势的情况下, 溶液中氧化还原对的费米能级高于p型半导体的费米能级, 电子会从溶液中转移到电极一侧, 这样电极一侧的空间电荷区会呈负电, 而溶液一侧则带正电.
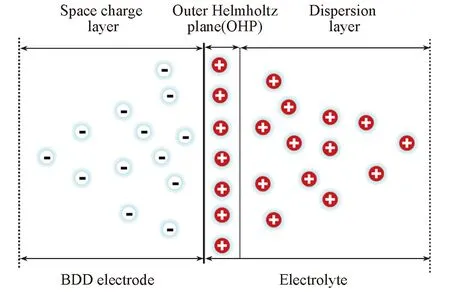
Fig.4 Schematic diagram of the interface between electrode and electrolyte
采用ZSimpWin3.10 软件, 选取适当的等效电路进行拟合并计算各部分电阻值. 这里对溶液一侧的双电层结构采用1个电容与1个电阻并联的等效电路进行拟合, 而半导体电极一侧的空间电荷层结构也采用1个电容和1个电阻并联的等效电路拟合, 考虑到溶液的电阻不能忽略, 经过反复尝试拟合, 在开路电位和1.0 V电位下, 按R(QR)(QR)的等效电路拟合, 而1.5 V电位由于有扩散引起的阻抗, 按R(Q(RZ\%W\%))(QR)等效电路拟合, 2.0和2.5 V电位下按R(QR)等效电路拟合. 选择的等效电路如图5所示, 拟合得到的连续曲线参见图3, 各参数的拟合值列于表1.
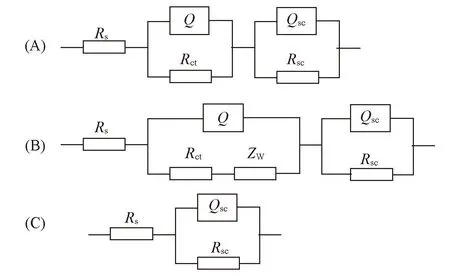
Fig.5 Equivalent circuit for electrochemical impedance spectroscopy(A) Open circuit and 1.0 V: Rs(QRct)(QscRsc); (B) 1.5 V: Rs(Q(RctZW))(QscRsc); (C) 2.0 and 2.5 V: Rs(QscRsc).
图5中Rs为溶液电阻;Rct为电荷转移电阻;ZW为韦伯阻抗;Q为常相位角元件, 对应溶液侧的双电层电容, 是用于描述双电层电容发生偏离的物理量;Qsc也为常相位角元件, 对应空间电荷层电容;Rsc为空间电荷层电阻.
从表1的拟合结果可知, 不同极化电位下溶液电阻Rs的平均值为(65.23±1.37) Ω·cm2, 在误差允许范围内基本相等. 在极化电位从开路电位增加至2,4-DCP的氧化峰电位1.5 V时, 电荷转移电阻Rct由4.275×104Ω·cm2降至2.885×102Ω·cm2, 表明提高电极电位有利于电催化氧化反应的进行, 可提高电催化反应速率. 在1.5 V电位条件下, 由于2,4-DCP氧化的电催化反应速率较快, 溶液中的2,4-DCP来不及扩散至电极表面, 并且2.4-DCP的氧化产物也未能及时扩散至溶液中, 因此反应速率由扩散步骤控制, 形成了韦伯阻抗. 而极化电位在2.0 V以上时, 高于BDD电极的析氧电位, 主要发生析氧反应, 使电催化反应的Rct降低为零, 因此在该电位值时Bode图(图2)只有1个峰值, 1个电容元件, 没有考虑拟合双电层. 发生析氧反应前, 开路电位至1.5 V时,Rsc值比较接近, 为(183.4±10.4) Ω·cm2. 当达到析氧电位以后,Rsc值迅速降低, 2.0和2.5 V时的值接近, 为(83.0±3.5) Ω·cm2, 表明析氧发生后空间电荷层的结构有所变化. 达到析氧电位后, 电极上至少同时发生2个反应, 一个是2,4-DCP在电极上的的直接电化学氧化反应, 另一个是H2O在电极上的电解反应, 产生·OH, 并进一步生成O2[16], 这可能是导致空间电荷层电阻降低的原因.

Table 1 Simulation results of BDD electrodes in 1 mmol/L 2,4-DCP+0.1 mol/L
*Rs: Solution resistance; Q-Yscand Q-nsc: parameters of constant phase element for space charge layer;Rsc: space charge layer resistance; Q-Y and Q-n: parameters of constant phase element for electric double layer;Rct: charge transfer resistance;Zw: Warburg resistance;Chisquared:Chi-Square goodness-of-fit test.
赵国华等[16]对苯酚在BDD电极上电化学阻抗谱的研究结果也表明, 在开路电位下, 在Bode图中出现2个峰, 说明有2个状态变量影响电极反应, 认为一个变量是电极电位(E), 另一个变量则是电极表面的某种状态变化量. 这与2,4-DCP在BDD电极上的阻抗谱类似. 但在苯酚氧化电位和析氧电位下, 阻抗谱的特征与本实验的结果不同, 这可能是由于2,4-DCP中—Cl基团的影响使得在电极上的氧化过程与苯酚不同.
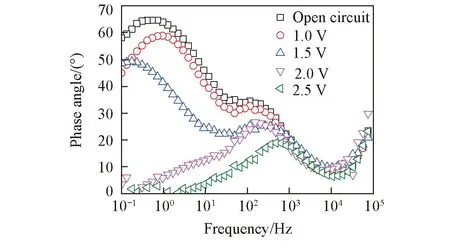
Fig.6 Bode plots of BDD electrodes in 1 mmol/L 2,6-DCP+0.1 mol/L Na2SO4 solution at different potentials
2.2.22,6-DCP的电化学阻抗谱电化学阻抗谱测试的溶液为1 mmol/L 2,6-DCP+0.1 mol/L Na2SO4, 其它测试条件与2.4-DCP相同, 得到的Bode图和Nyquist图如图6和图7所示.
从图6和图7可看出, 2,6-DCP的电化学阻抗谱与2,4-DCP相似, 在开路电位、1.0 V和1.5 V电位下, 相位角对频率的曲线有2个峰, 推测至少有2个电容元件; 而2.0和2.5 V的曲线仅有1个峰, 推测至少有1个电容元件. 由1.5 V的Nyquist曲线可以看出, 高频有1个容抗弧, 低频曲线为近似呈45°角的直线, 说明有扩散引起的韦伯阻抗. 同样, 开路电位和1.0 V电位下, 按R(QR)(QR)的等效电路拟合, 而1.5 V电位按R(Q(RZ\%W\%))(QR)等效电路拟合, 2.0和2.5 V电位下按R(QR)等效电路拟合, 选择的等效电路如图5所示, 拟合得到的连续曲线见图7, 各参数的拟合值列于表2.

Fig.7 Nyquist plots of BDD electrodes in 1 mmol/L 2,6-DCP+0.1 mol/L Na2SO4 solution at different potentials (A) Open circuit potential; (B) 1.0 V; (C) 1.5 V; (D) 2.0 V; (E) 2.5 V.

Potential/VRs/(Ω·cm2)105Q-Ysc/(S·sn·cm2)Q-nscRsc/(Ω·cm2)104Q-Y/(S·sn·cm2)Q-nRct/(Ω·cm2)104ZW/(S5s0.5·cm2)104ChisquaredOpencircuit80.094.1900.7480235.70.9150.81265.463×1043.5241.080.014.1680.7464228.81.1430.80491.981×1044.3121.579.074.4280.6757270.76.3300.87094.972×1037.7537.9752.077.3915.7200.5278483.546.892.571.236.0290.6632154.613.48
3结论
采用循环伏安法和电化学阻抗谱对BDD电极上2,4-DCP和2,6-DCP的电催化氧化过程进行了研究, 并利用等效电路对氯酚的电化学阻抗谱进行了拟合, 得到不同极化电位下电化学元件相应的参数值. 研究发现, 2,4-DCP和2,6-DCP的氧化电位约为1.55和1.62 V, 当极化电位由开路电位提高至氧化电位1.5 V时, 2种氯酚的电荷转移电阻均有明显下降, 说明提高电极电位有利于电催化氧化反应进行, 且氧化电位下的反应受扩散步骤控制. 通过比较发现, 2,4-DCP在BDD电极上比2,6-DCP更容易被直接电化学氧化.
参考文献
[1]Carey J. J., Christ J. C. S., Lowery S. N.,MethodofElectrolysisEmployingADopedDiamondAnodetoOxidizeSolutesinWastewater, US 5399247, 1995-03-21
[2]Troster I., Fryda M., Herrmann D., Schafer L., Hanni W., Perret A., Blaschke M., Kraft A., Stadelmann M.,DiamondRelat.Mater., 2002, 11(3—6SI), 640—645
[3]Moreira F.C., Garcia-Segura S., Boaventura R., Brillas E., Vilar V.,Appl.Catal.B.,Environ., 2014, 160, 492—505
[4]Groenen-Serrano K., Weiss-Hortala E., Savall A., Spiteri P., Electrocatalysis, 2013, 4(4), 346—352
[5]Iniesta J., Michaud P. A., Panizza M., Cerisola G., Aldaz A., Comninellis C., Electrochim. Acta, 2001, 46(23), 3573—3578
[6]Zhi J. F., Tian R. H., Process Chem., 2005, 17(1), 55—63(只金芳, 田如海. 化学进展, 2005, 17(1), 55—63)
[7]Wang S. S., Chen G. H., Yang F. L., Thin Solid Films, 2009, 517(12), 3559—3561
我立刻起身来到小唐身边,牵着他的手走到教室外的阳光下。“你有什么愿望没有实现吗?需要我的帮助吗?”一听我的话,小唐的眼泪又流了下来,他边哭边说:“中午我帮同学们打饭,同学不让我帮忙,说我打饭不安全。”看着小唐的脸,听着小唐的话,我的心中真的很感动。我很能理解小唐的话,也很能理解小唐的心。
[8]Panizza M., Cerisola G., Electrochim.Acta, 2005, 51(2), 191—199
[9]Habka N., Pinault M. A., Mer C., Jomard F., Barjon J., Nesladek M., Bergonzo P., Phys. Status Solidi A, 2008, 205(9), 2169—2172
[10]Liang L. Q., Huang W. M., Lin H. B., Chem. J. Chinese Universities, 2015, 36(8), 1606—1611(梁龙琪, 黄卫民, 林海波. 高等学校化学学报, 2015, 36(8), 1606—1611)
[11]Tian Y., Chen X. M., Shang C., Chen G. H., J. Electrochem. Soc., 2006, 153(7), J80—J85
[12]Huang W. M., Lin H. B., Chem. J. Chinese Universities, 2015, 36(9), 1765—1770(黄卫民, 林海波. 高等学校化学学报, 2015, 36(9), 1765—1770)
[13]Zhi J. F., Wang H. B., Nakashima T., Rao T. N., Fujishima A., J. Phys.Chem. B, 2003, 107(48), 13389—13395
[14]Lv J. W., Feng Y. J., Liu J. F., Qu Y. P., Cui F. Y., Appl. Surf. Sci., 2013, 283, 900—905
[15]Xu J. S., Granger M. C., Wang J., Chen Q. Y., Witek M. A., Hupert M. L., Hanks A., Swain G. M., Sakaguchi I., Nishitani-Gamo M., Ando T., Diamond Materials VI, 2000, 99(32), 403—415
[16]Zhao G. H., Li M. L., Qi Y., Hu H. K., China Environ. Sci., 2005, 25(3), 370—374(赵国华, 李明利, 祁源, 胡惠康. 中国环境科学, 2005, 25(3), 370—374)
[17]Zhao G. H., Li M. L., Wu W. W., Li R. B., He X. C., Environ. Sci., 2004, 25(5), 163—167(赵国华, 李明利, 吴薇薇, 李荣斌, 何贤昶. 环境科学, 2004, 25(5), 163—167)
[18]Liu F. B., Li X. M., Wang J. D., Liu B., Chen D. R., Chinese Sci. Bull., 2006, 51(11), 1344—1348(刘峰斌, 李学敏, 汪家道, 刘兵, 陈大融. 科学通报, 2006, 51(11), 1344—1348)
[19]Cui K., Wang J. D., Feng D., Chen D. R., J. Funct. Mater., 2015, 46(7), 7076—7080(崔凯, 汪家道, 冯东, 陈大融. 功能材料, 2015, 46(7), 7076—7080)
[20]Feng Y. J., Lv J. W., Liu J. F., Gao N., Peng H. Y., Chen Y. Q., Appl. Surf. Sci., 2011, 257(8), 3433—3439
[21]Zha Q. X., Introduction to Kinetics of Electrode Process, Science Press, Beijing, 2002, 385(查全性. 电极过程动力学导论, 北京: 科学出版社, 2002, 385)
Electrochemical Impedance Spectroscopy of Dichlorophenols
at Boron-doped Diamond Electrodes†
LÜ Jiangwei1*, QU Youpeng2, FENG Yujie3, LIU Junfeng3
(1.SchoolofPharmacy,HarbinUniversityofCommerce,Harbin150076,China;
2.SchoolofLifeScienceandTechnology,HarbinInstituteofTechnology,Harbin150080,China;
3.StateKeyLaboratoryofUrbanWaterResourceandEnvironment,HarbinInstituteofTechnology,Harbin150090,China)
AbstractCyclic voltammetry(CV) and electrochemical impedance spectroscopy(EIS) were used to investigate the electrocatalytic process of boron-doped diamond(BDD) electrode with 2,4-dichlorophenol(2,4-DCP) and 2,6-dichlorophenol(2,6-DCP) as target pollutants. Results showed that the oxidation potential of the 2,4-DCP and 2,6-DCP were 1.55 and 1.62 V, respectively. Equivalent circuit simulation results showed that the charge transfer resistance for the two DCPs were both decreased when the polarization potential increased from open circuit potential to 1.5 V, indicating that electrocatalytic oxidation was favored by increasing potential. The diffusion step was the control step at oxidation potential. The indirect electrochemical oxidation of 2,4-DCP occurred more easily at BDD electrode than 2,6-DCP.
KeywordsBoron-doped diamond; Cyclic voltammetry; Electrochemical impedance spectroscopy; Dichlorophenol; Electrocatalysis
(Ed.: S, Z, M)
† Supported by the National Natural Science Foundation of China(Nos.51308171, 51209061), the Science and Technology Project of Heilongjiang Province Education Department, China(No.12541185) and the Fundamental Research Fund for the Central Universities, China(No.HIT.NSRIF.2015090).

doi:10.7503/cjcu20150696
基金项目:国家自然科学基金(批准号: 51308171, 51209061)、黑龙江省教育厅科学技术研究项目(批准号: 12541185)和中央高校基本科研业务费专项基金(批准号: HIT.NSRIF.2015090)资助.
收稿日期:2015-05-08. 网络出版日期: 2015-12-20.
中图分类号O646
文献标志码A
联系人简介: 吕江维, 女, 博士, 讲师, 主要从事环境电化学研究. E-mail: pp198259@163.com
