基于链置换循环无标记检测端粒酶RNA的荧光法
2016-04-08张霞菲时志路
张霞菲, 成 锐, 时志路, 金 燕
(陕西师范大学化学化工学院, 陕西省生命分析重点实验室, 陕西 710062)
基于链置换循环无标记检测端粒酶RNA的荧光法
张霞菲, 成锐, 时志路, 金燕
(陕西师范大学化学化工学院, 陕西省生命分析重点实验室, 陕西 710062)
摘要以氧化石墨烯(GO)作为DNA载体和荧光猝灭剂, SYBR Green Ⅰ(SGⅠ)为荧光信号探针, 发夹核酸探针为分子识别探针, 基于目标物启动的发夹核酸探针链置换循环反应, 建立了一种利用荧光共振能量转移和链置换循环放大技术检测端粒酶RNA(hTR)的荧光新方法. 发夹核酸探针hpDNA1和hpDNA2吸附在GO表面, 嵌插在发夹DNA探针茎部的SGⅠ的荧光信号被GO猝灭. 当人工合成的目标物(T1)存在时, T1与hpDNA1杂交打开hpDNA1的茎-环结构而引发hpDNA2与T1之间的链置换循环反应, 由此累积产生大量的hpDNA1/hpDNA2杂交双链. 刚性的双链DNA脱离GO表面, 导致所嵌插的SGⅠ产生较强的荧光信号. 基于荧光信号的变化, 可定量检测0.2~50 nmol/L的T1, 检出限为90 pmol/L. 该方法为端粒酶RNA检测提供了一种高灵敏、高特异性且无需标记的荧光新途径.
关键词端粒酶RNA; 链置换循环; 荧光共振能量转移; 无标记
E-mail: jinyan@snnu.edu.cn
端粒酶(Telomerase)是一种核糖核蛋白酶, 其主要功能是催化端粒(Telomere)末端不断延伸(TTAGGG)n序列, 从而克服细胞分裂过程中的端粒缩短问题. 研究表明, 端粒酶在85%以上的人类肿瘤细胞中被高表达, 而在正常体细胞中的表达量则极低甚至不表达. 因此, 端粒酶与细胞的恶性转化及持续分裂增殖具有密切关系, 是恶性肿瘤发展程度的衡量指标[1~8]. 端粒酶由3个主要成分组成, 其中端粒酶RNA(hTR)是端粒酶活性表达的核心组件[9~11], 它是长为451 bp 的核苷酸短片段, 含有长11个碱基的模板区域(5′-CUAACCCUAAC-3′), 在正常细胞和肿瘤细胞中都有表达.
近年来的研究[12,13]表明, hTR在多种肿瘤中有异常的扩增现象, 与正常组织相比表达水平偏高, 显示出其在致肿瘤性上的重要作用. 目前, 检测hTR的方法主要有原位杂交法[14,15]和逆转录PCR[16,17]法等. Shay等[18]采用原位杂交技术检测了食道癌细胞中RNA的含量. Naoki等[19]采用实时荧光定量法检测了hTR的表达水平. Gao等[20]以银纳米簇作为信号分子建立了检测hTR的荧光分析法. 这些方法存在操作较为繁琐和灵敏度不高等局限. 因此, 亟需发展一种简便、灵敏检测hTR的新方法.
近年来, 信号放大技术[21,22]备受关注, 尤其是链置换信号放大技术作为一种等温、无酶辅助的新型放大技术, 被广泛用于检测DNA、蛋白质、小分子及金属离子等目标物. 采用2个发夹核酸探针, 通过目标物启动的链置换循环反应, 在无酶的条件下可实现目标物的高灵敏检测.
本文以氧化石墨烯(GO)为DNA载体和荧光猝灭剂, 以发夹核酸探针为分子识别探针, 基于GO对发夹核酸探针和双链DNA吸附能力的差异, 将目标物引起的荧光共振能量转移变化和目标物启动的链置换循环反应相结合, 建立了一种在均相溶液中简便、快速且无需标记检测hTR的荧光新方法.
1实验部分
1.1试剂与仪器
氧化石墨烯(GO, 南京先锋纳米材料科技有限公司); SYBR Green Ⅰ[SGI, 美国NEB(Life Technologies)公司]; MgCl2(西安化学试剂厂); 三羟甲基氨基甲烷(Tris, 北京鼎国生物科技有限公司); 实验所用DNA序列如表S1(见本文支持信息)所示, 均由上海生物工程有限公司合成, 用20 mmol/L Tris-HCl(pH=7.4)和5 mmol/L MgCl2缓冲溶液配制成100 μmol/L的储备液, 置于冰箱中备用. 实验用水均为超纯水(18.2 MΩ·cm).
Hitachi F-7000型荧光分光光度计(日本日立公司); LS型超纯水机(美国PALL公司); 5424R型小型离心机(德国Eppendorf公司); PowerPac Basic型电泳仪(美国伯乐BIO-RAD公司); BIO-RAD型凝胶成像系统(上海培清科技有限公司); WFH-204B型手提式紫外灯(上海精科实业); IX73型倒置荧光显微镜(日本Olympus公司); iXon X3 885 型电子倍增电荷耦合装置(EMCCD, 英国Andor公司).
1.2实验过程
1.2.1荧光测定向20 nmol/L hpDNA1中加入一定量的SGⅠ和GO, 在20 mmol/L Tris-HCl(pH=7.4)和5 mmol/L MgCl2缓冲溶液中培育10 min, 测定其荧光强度. 加入不同浓度的目标物, 再测定荧光强度. 进行链置换循环放大时, 在hpDNA1/SGⅠ/GO混合物中加入不同浓度的目标物和一定量的hpDNA2, 在室温下培育20 min, 测定荧光强度. hpDNA1, hpDNA2, GO 和SGⅠ的最终浓度分别为20 nmol/L, 30 nmol/L, 15 μg/mL和6 μL 20×SGⅠ.
1.2.2比色法测定将1 μmol/L hpDNA1, 1.5 μmol/L hpDNA2, 1 μmol/L T1, 5 μL 50×SGⅠ 和50 μg/mL GO在20 mmol/L Tris-HCl(pH=7.4)和5 mmol/L MgCl2缓冲溶液中反应1 h后, 在暗室中用紫外灯照射并拍照.
1.2.3凝胶电泳分析将100 nmol/L hpDNA1, 150 nmol/L hpDNA2和100 nmol/L目标物T1混合于20 mmol/L Tris-HCl(pH=7.4)和5 mmol/L MgCl2缓冲溶液中, 培育30 min. 向每10 μL的样品中加入1 μL染色液[0.25%(质量分数)溴酚蓝+0.25%(质量分数)二甲苯蓝(FF)+30%(质量分数)甘油]充分混合, 取2 μL混合液上样进行电泳分析. 凝胶电泳在12.5%(质量分数)的聚丙烯酰胺凝胶中进行, 电泳液为1×TBE缓冲液(90 mmol/L Tris硼酸盐+1 mmol/L EDTA, pH=8.3), 于200 V电压下电泳20 min. 电泳结束后用硝酸银染色, 再用凝胶成像仪成像.
1.2.4细胞培养将宫颈癌细胞(HeLa)培养于含10%(体积分数)胎牛血清的DMEM培养基中(含有100 U/mL青霉素+0.01 mg/mL链霉素+10%胎牛血清), 传代后在37 ℃, 95%(体积分数)空气和5% CO2条件下培养, 直至贴壁细胞的丰度>60%.
1.2.5GO对核酸保护作用的分析将100 nmol/L hpDNA1与10U DNase 1 混合, 分别培育0, 5, 10, 30和60 min, 于95 ℃失活5 min后进行凝胶电泳分析. 同时, 将100 nmol/L hpDNA1, 10U DNase 1和15 μg/mL GO 培育后进行凝胶电泳分析.
1.2.6GO对细胞毒性的分析首先将Hela细胞以每孔104个细胞接种在96孔板中, 待细胞贴壁后加入不同浓度的GO并作用8 h, 用PBS缓冲液清洗, 再加入20 μL MTT(5 mg/mL)作用4 h后用二甲基亚砜(DMSO)溶解, 采用酶标仪测定490 nm处的光密度OD值, 进行细胞毒性分析.
1.2.7细胞中hTR的荧光成像分析将Hela细胞栽种在35 mm培养皿中, 用DMEM 培养基培育至细胞贴壁. 将F-hpDNA1/GO混合液和F-hpDNA1/hpDNA2/GO混合溶液分别与Hela 细胞进行培育. 核酸探针的最终浓度为100 nmol/L F-hpDNA1和150 nmol/L hpDNA2, GO的最终浓度为15 μg/mL. 培育 6~8 h后, 用 PBS 缓冲液反复洗涤细胞, 最后加入适量PBS 缓冲溶液进行显微镜分析.
2结果与讨论
2.1实验原理
基于荧光共振能量转移和链置换循环放大技术建立的hTR表达水平检测原理如Scheme 1所示. 该方法以GO作为荧光猝灭剂, 以发夹核酸探针作为分子识别探针, SGⅠ作为荧光信号分子. SGⅠ自身荧光信号很微弱, 当其嵌插到核酸探针磷酸骨架的双链区则会产生强荧光信号. 当不存在目标物T1时, 发夹核酸探针hpDNA1和hpDNA2可以稳定共存, 通过5′黏性末端和较大的环部序列π-π堆积作用吸附到GO的表面, 使得嵌插于发夹核酸探针茎部双链区的SGⅠ与GO靠近, 发生荧光共振能量转移(FRET), SGⅠ的荧光信号被猝灭. 而当存在目标物T1时, T1与hpDNA1悬垂以及茎部杂交破坏发夹结构, 释放出的长单链部分序列可打开hpDNA2, 形成的hpDNA1/hpDNA2杂交双链DNA可有效置换出目标物T1启动链置换反应, 最终产生大量的hpDNA1/hpDNA2杂交双链. 刚性较强的杂交双链完全脱离GO, 使得嵌插的SGⅠ产生强荧光信号. 链置换循环反应实现了荧光信号的累积放大, 基于此构建了无标记检测hTR的荧光新方法.
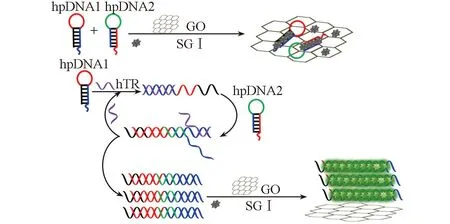
Scheme 1 Schematic illustration of a label-free strand displacement amplification strategy for sensitive detection of hTR
2.2原理验证
2.2.1荧光和比色分析为了验证检测原理的可行性, 首先进行了荧光分析. 如图1所示, SGⅠ自身荧光信号很微弱, 当其嵌插到发夹核酸探针茎部的双链区时产生较强的荧光信号. GO存在时, 发夹核酸探针吸附到GO表面, SGⅠ荧光信号被GO猝灭. 当目标物T1存在时, hpDNA1被打开, 双链DNA从GO表面脱落, GO与SGⅠ间的荧光共振能量转移效率减弱, SGⅠ荧光信号部分恢复. 加入hpDNA2后, 打开的hpDNA1与 hpDNA2杂交生成更长更稳定的双链DNA, 并释放出T1进行循环反应, 生成更多的双链DNA, 能够嵌插大量的SGⅠ, 导致荧光信号显著增强. 此结果初步证明了原理的可行性.

Fig.1 Fluorescence emission spectra of SGⅠ(A) a. SGⅠ; b. SGⅠ+hpDNA1+hpDNA2; c. SGⅠ+hpDNA1+hpDNA2+GO; d. SGⅠ+hpDNA1+hpDNA2+GO+T1. Inset of (A): photographs under UV light. a. SGⅠ+hpDNA1+hpDNA2; b. SGⅠ+htDNA1+hpDNA2+GO; c. SGⅠ+hpDNA1+T1; d. SGⅠ+hpDNA1+hpDNA2+GO+T1.

Fig.2 Gel electrophoretic analysis of strand displacement amplification strategyLane M: marker; lane 1: hpDNA1; lane 2: hpDNA2; lane 3: T1; lane 4: hpDNA1+T1; lane 5: hpDNA2+T1; lane 6: hpDNA1+hpDNA2; lane 6: hpDNA1+hpDNA2+T1.
嵌插于DNA杂交双链中的(SGⅠ)经紫外灯照射能产生肉眼可见的绿色荧光, 通过比较紫外灯照射下目标物引入前后体系颜色的变化, 建立了目标物T1检测的比色法. 如图1(A)插图a与b所示, hpDNA1和hpDNA2可以吸附到GO表面, SGⅠ荧光信号被猝灭; 而c与d结果显示, 链置换循环放大反应的发生致使荧光信号明显增强. 比色分析结果与荧光检测结果一致, 进一步证明了原理的可行性. 为了排除其它因素引起的荧光信号变化, 设计了负对照实验. 如图1(B)所示, 在SGⅠ/hpDNA1/hpDNA2/GO混合液中, 加入任意序列的DNA(T4), SGⅠ荧光信号强度几乎无变化, 说明只有在目标物存在的情况下, 荧光信号才会得到增强, 进一步证明了该方法的可靠性.
2.2.2凝胶电泳分析凝胶电泳可有效验证DNA的构型变化, 结果如图2所示. 图2泳道4中出现了1条新的条带, 且T1带消失, 说明T1能够与hpDNA1进行杂交. 泳道5结果表明, T1不会直接与hpDNA2杂交, 二者可以稳定存在. 泳道6中hpDNA1与hpDNA2混合后没有新的条带形成, 说明在没有目标物存在的情况下, 2个发夹DNA可以各自稳定存在, 不会相互打开. 当存在目标物时, 泳道7中不仅观察到了hpDNA1/T1杂交带, 而且还观察到了hpDNA1/hpDNA2的杂交带, 说明目标物存在下hpDNA2可以有效地与hpDNA1杂交, 并替代目标物T1进行循环反应, 以上电泳结果充分说明该方法的可行性与可靠性.
2.3实验条件的优化
为了获得最佳的实验效果, 对一些重要条件进行了优化, 优化过程及结果如图S1~图S5(见本文支持信息)所示. 实验选择氧化石墨的烯浓度为15 μg/mL, hpDNA1与hpDNA2的浓度比为1∶1.5, SGⅠ用量为6 μL 20×SGⅠ, 反应温度为37 ℃, 于pH=7.4的缓冲溶液中反应20 min作为目标物T1检测的最佳条件.
2.4方法的选择性
为了考察本方法对目标物T1的识别选择性, 分别研究了hpDNA1/hpDNA2/GO混合溶液与目标物(T1)、单碱基错配的DNA(T2)、2个碱基错配的DNA(T3)和完全不互补的DNA(T4)的作用情况. 如图3所示, 在相同的实验条件下, 这4种DNA序列检测的荧光信号存在明显的差异, 含有T1的体系荧光信号明显强于其它DNA所产生的信号. 图3中插图为比色分析结果, a~e与下方的柱形图依次对应, 可见, 经紫外灯照射后, 含有T1的溶液产生的光可明显区别于其它DNA分子产生的光. 综上, 荧光分析和比色分析结果均表明该方法具有较好的选择性.
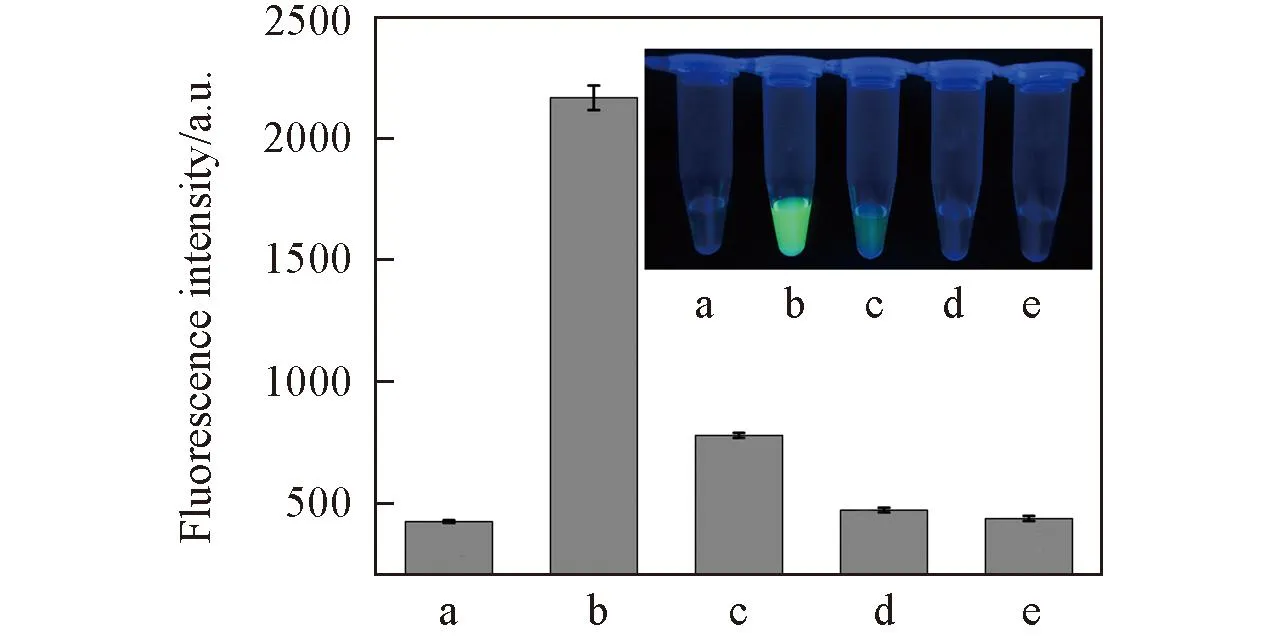
Fig.3 Investigation of the selectivity of the proposed methoda. SGⅠ+hpDNA1+hpDNA2/GO; b. SGⅠ+HPDNA1+hpDNA2+T1; c. SGⅠ+hpDNA1+hpDNA2+single-base mismatched DNA; d. SGⅠ+hpDNA1+hpDNA2+two-base mismatched DNA; e. SGⅠ+hpDNA1+hpDNA2+completely mismatch DNA. Inset: photographs under UV light.
2.5目标物T1的检测灵敏度

Fig.4 Fluorescence emission spectra of SGⅠ/hpDNA1/hpDNA2 in the presence of T1(A) and the linear relationship between the fluorescence intensity and the concentrations of T1(B) (A) c(T1)/(nmol·L-1), a—f: 0, 10, 20, 30, 40, 50.
对最佳条件下不含有hpDNA2时的方法灵敏度进行了考察. 由图4(A)可见, 随着目标物T1浓度的增加, hpDNA1/GO/SGⅠ体系的荧光强度逐渐增强. 在10~50 nmol/L范围内, 体系的荧光强度与T1浓度值呈现良好的线性关系, 相关系数为0.9976[图4(B)], 检出限为5.50 nmol/L(n=11).
对最佳条件下含hpDNA2时方法灵敏度的考察结果如图5所示. 由图5(A)可见, 随着目标物T1浓度的增加, hpDNA1hp/hpDNA2/GO/SGⅠ体系的荧光强度逐渐增强. 检测范围为0.2~50 nmol/L, 且在0.2~1 nmol/L 范围内荧光强度与hTR浓度呈现较好的线性关系, 相关系数为0.9957, 检出限为90 pmol/L[图5(B)]. 以上结果表明, hpDNA2的引入提高了目标物检测的灵敏度.

Fig.5 Fluorescence emission spectra of GO/SGⅠ/hpDNA1/hpDNA2 in the presence of T1(A) and the linear relationship between the fluorescence intensity and concentrations of T1(B) (A) c(T1)/(nmol·L-1), a—m: 0, 0.2, 0.4, 0.6, 0.8, 10, 2, 5, 10, 20, 30, 40, 50. Inset of (B) is the linear relationship in T1 concentration range of 0.2—1.0 nmol/L.
2.6GO对核酸的保护作用

Fig.6 Gel electrophoresis analysis of hpDNA1 treated with DNase 1 in the absence and presence of GO for 5, 10, 30 and 60 min
如何防止核酸降解是检测实际样品中核酸或者脱氧核酸面临的最大挑战. GO不仅可作为荧光猝灭剂, 还可保护核酸, 防止其被酶降解. 以hpDNA1为模式DNA分子, DNase1为核酸降解酶, 它能够消化单链或者双链DNA产生单脱氧核苷酸. 如图6所示, 在不含GO的体系中, 10 min内hpDNA1几乎被完全降解; 而在含有GO的体系中, hpDNA1的量几乎无变化. 此结果说明GO对核酸起到了保护作用, 该方法可用于复杂样品或细胞中目标物的检测.
2.7GO对细胞的毒性
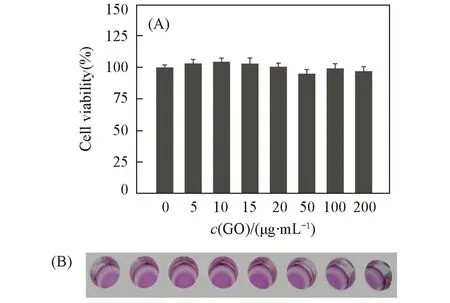
Fig.7 Cytotoxicity test of the GO in HeLa cell(A)and the color change of formazan crystals with different concentrations of GO(B)
为了利用GO将链置换反应的探针送入细胞内, 首先利用噻唑蓝(MTT)法考察了GO对HeLa细胞的毒性. MTT为黄色化合物, 是一种接受氢离子的染料, 可作用于活细胞线粒体中的呼吸链, 在琥珀酸脱氢酶和细胞色素C的作用下生成蓝色(或蓝紫色)不溶于水的甲臜结晶, 甲臜结晶的生成量仅与活细胞数目成正比(死细胞中琥珀酸脱氢酶消失, 不能将MTT还原). 还原生成的甲臜结晶可用二甲基亚砜(DMSO)溶解, 利用酶标仪测定490 nm处的光密度OD值, 即可反映活细胞数目, 从而进行细胞毒性分析.
图7(A)为不同浓度的GO与细胞作用 8 h 后所对应的细胞存活率, 图7(B)对应图7(A)中每个GO浓度下MTT结果的颜色变化. 可以看出, 随着GO浓度的增大, 细胞存活率并未发生明显降低, 处于一个相对稳定的状态, 当GO浓度达到 200 μg/mL时, 细胞存活率仍高达 96.96%. 因此, 可以认为GO对细胞无明显毒性, 可作为生物载体实现细胞内目标物质的检测.
2.8细胞内hTP的荧光成像分析
对hpDNA1的5′端进行6-羧基荧光素(FAM)标记(记作F-hpDNA1), 用于测定细胞中hTR含量. 基于活细胞对纳米尺度材料进行细胞自吞噬的机理, 将F-hpDNA1/hpDNA2/GO纳米复合物与HeLa细胞共培养, 实现了基于GO对链置换反应探针的细胞跨膜运输, 结果如图8所示. 图8(A1)~(A3)示出了GO携带/F-hpDNA1进入细胞的结果, hTR将F-hpDNA1打开, 发出微弱的荧光信号. 图8(B1)~(B3)示出了GO携带F-hpDNA1/hpDNA2进入细胞的结果, 其荧光强度强于(A1)~(A3)的荧光强度, 说明hpDNA2的引入致使细胞中发生链置换循环反应, 荧光信号明显增强. 该结果也说明本文方法的原理是可行的, 同时进行标记还可用于细胞中hTR的检测.

Fig.8 Intracelluar images of HeLa cells incubated with GO/F-hpDNA1(A1—A3) and GO/F-hpDNA1/hpDNA2 for 8 h(B1—B3)(A1), (B1): Bright-field images; (A2), (B2): fluorescence images; (A3), (B3): merge of fluorescence and bright-field images.
综上所述, 基于氧化石墨烯的FRET效应, 以荧光染料为信号分子, 提出了一种基于链置换循环放大技术的灵敏、快速无标记检测hTR的荧光新方法. 以2个发夹核酸探针在目标序列存在下自组装设计的链置换循环放大法无需酶辅助, 反应时间短, 检测效率高. 该方法采用SGⅠ作为信号分子, 降低了实验成本, 也可通过紫外灯照射进行可视化分析. 以GO作为输送载体和荧光猝灭剂, 可通过细胞内成像实现细胞中hTR的检测. 对目标物的灵敏检测以及对单碱基错配序列的识别说明该方法有高的灵敏度和选择性. 该方法为hTR的检测提供了一种高选择性、无需标记且低成本的荧光新途径.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20150524.
参考文献
[1]Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M.,Science, 1994, 266, 2011—2015
[2]Hiyama E., Yokoyama T., Tatsumoto N., Hiyama K., Imamura Y., Murakami Y., Kodama T., Piatyszek M.A., Shay J. W., Matsuura Y.,CancerRes., 1995, 55, 3258—3262
[3]Hiyama E., Gollahon L., Kataoka T., Kuroi K., Yokohama T., Gazder A. F., Hiyama K., Piatyszek M.A., Shay J.W.,J.Natl.CancerInst., 1996, 88, 116—122
[4]Lin Y., Miyamoto H., Fujinami K., Uemura H., Hosaka M., Iwasaki Y., Kubota Y.,Clin.CancerRes., 1996, 2, 929—932
[5]Chadeneau C., Hay K., Hirte H. W., Gallinger S., Bacchetti S.,CancerRes., 1995, 55, 2533—2563
[6]Sommerfeld H. J., Meeker A. K., Piatyszek M. A., Bova G. S., Shay J. W., Coffey D. S.,CancerRes., 1996, 56, 218—222
[7]Yu G. L., Bradley J. D., Attardi L. D., Blackburn E. H.,Nature, 1990, 344, 126—132
[8]Fan H. L., Zhang T., Jin W., Jin Q. H.,Chem.J.ChineseUniversities, 2015, 36(4), 625—632(范宏亮, 张涛, 金伟, 金钦汉. 高等学校化学学报, 2015, 36(4), 625—632)
[9]Feng J., Funk W. D., Wang S. S., Weinrich S. L., Avilion A. A., Chiu C. P., Adams R. R., Chang E., Allsopp R. C., Yu J. H., Le S., West M. D., Harley C. B., Andrews W. H., Greider C. W., Villeponteau B.,Science, 1995, 269, 1236—1241
[10]Counter C. M., Meyerson M., Eaton E. N., Ellisen L. W., Caddle S. D., Haber D. A., Weinberg R. A.,Oncogene, 1998, 16, 1217—1222
[11]Blackburn E. H.,Mol.CancerRes., 2005, 3, 477—482
[12]Shay J. W., Bacchetti S.,Eur.J.Cancer, 1997, 33, 787—791
[13]Counter C. M., Avilion A. A., Lefeuvre C. E., Stewart N. G., Greider C. W., Harley C. B., Bacchetti S.,J.EMBO, 1992, 11, 1921—1929
[14]Yashima K., Piatyszek M. A., Saboorian H. M., Virmani A. K., Brown D., Shay J. W., Gazdar A. F.,J.Clin.Pathol., 1997, 50, 110—117
[15]Yashima K., Ashfaq R., Nowak J., Gruenigen V. V., Milchgrub S., Rathi A., Albores-Saavedra J.,Cancer, 1998, 82, 1319—1327
[16]Heine B., Hummel M., Demel G., Stein H.,J.Pathol., 1999, 188, 139—145
[17]Zhang L. H., Zhuo. T.,Sci.ChinaChem., 2014, 57, 961—965
[18]Morales C. P., Lee E. L., Shay J. W.,Cancer, 1998, 83, 652—659
[19]Tomomi Y., Atsuhito Y., Hidekazu K., Daisuke K., Daisuke F., Koichi H., Naoki W.,Clin.Chem., 1998, 44, 2441—2445
[20]Wei Y. T., Liu R., Sun Z. P., Wang Y. L., Cui Y. Y., Zhao Y. L., Cai Z. F., Gao X. Y.,Analyst, 2013, 138, 1338—1341
[21]Yang H. W., Liang W. B., Si J., Li Z. Y., He N. Y.,J.Biomed.Nanotechnol., 2014, 10, 3610—3619
[22]Yang H. W., Liang W. B., He N. Y., Deng Y., Li Z. Y.,ACSAppl.Mater.Interfaces, 2015, 7, 774—781
Label-free Fluorescence Assay of Telomerase RNA Based on
Strand Displacement Amplification†
ZHANG Xiafei, CHENG Rui, SHI Zhilu, JIN Yan*
(AnalyticalChemistryforLifeScienceofShaanxiProvince,SchoolofChemistryand
ChemicalEngineering,ShaanxiNormalUniversity,Xi’an710062,China)
AbstractA novel fluorescence method was developed for detecting telomerase RNA(hTR) based on the fluorescence resonance energy transfer(FRET) and strand displacement amplification(SDA) technique. Graphene oxide(GO) served as DNA carrier and fluorescence quencher. SYBR Green Ⅰ(SGⅠ) and hairpin DNAs(hpDNA) are fluorescence probe and molecular recognition probes, respectively. The fluorescence of SGⅠ that intercalated into the stem of hairpin DNAs was quenched when hpDNA1 and hpDNA2 were adsorbed onto the surface of GO. In the presence of T1, the hybridization reaction between hpDNA1 and T1 opened the hairpin structure of hpDNA1 to trigger the SDA reaction between hpDNA2 and T1, leading to an accumulation of hpDNA1/hpDNA2 hybrids. The rigid dsDNA desorbed from GO surface to restore the fluorescence of SGⅠ. Based on the change in fluorescence intensity, T1 can be quantitatively detected from 0.2 nmol/L to 50 nmol/L, with a detection limit of 90 pmol/L. Therefore, it offers a label-free, highly sensitive and specific fluorescence strategy for detection of hTR.
KeywordsTelomerase RNA; Strand displacement amplification; Fluorescence resonance energy transfer; Label-free
(Ed.: N, K)
† Supported by the National Natural Science Foundation of China(Nos.21075079, 21375086), the Program for Innovative Research Team in Shaanxi Province, China(No.2014KCT-28), the Fundamental Research Funds for the Central Universities, China(No.GK261001097) and the Program for Changjiang Scholars and Innovative Research Team in University, China(No.IRT-14R33).
doi:10.7503/cjcu20120521
基金项目:国家自然科学基金(批准号: 21075079, 21375086)、陕西省创新团队研究计划项目(批准号: 2014KCT-28)、陕西师范大学中央高校基金项目(批准号: GK261001097)和长江学者高校创新团队项目(批准号: IRT-14R33)资助.
收稿日期:2015-07-08. 网络出版日期: 2015-12-20.
中图分类号O657.3
文献标志码A
联系人简介:金燕, 女, 博士, 教授, 博士生导师, 主要从事生物传感及生化分析方面的研究.
