异质结型AgBr/CuO光催化剂的合成、光催化活性及再生
2016-04-08刘建新王雅文樊彩梅段东红王韵芳
张 雪, 刘建新, 王雅文, 樊彩梅, 段东红, 王韵芳
(太原理工大学洁净化工研究所, 太原 030024)
异质结型AgBr/CuO光催化剂的合成、光催化活性及再生
张雪, 刘建新, 王雅文, 樊彩梅, 段东红, 王韵芳
(太原理工大学洁净化工研究所, 太原 030024)
摘要采用沉淀法制备了具有p-n异质结结构的AgBr/CuO可见光催化剂, 对其结构进行了表征, 通过甲基橙溶液的降解率评价了AgBr/CuO的光催化活性, 并通过活性物种测试及能带结构分析推测了其光催化机理, 采用3%(质量分数)溴水对使用后的AgBr/CuO进行了再生处理. 结果表明, 在可见光照射下, 0.1 g AgBr/CuO光催化剂30 min对甲基橙溶液(初始浓度为15 mg/L)的降解率高达92%, 远高于同等条件下的AgBr. AgBr/CuO光催化活性提高的原因是AgBr与CuO的复合一方面使催化剂的禁带宽度变宽, 提高了光生电子与光生空穴的氧化还原能力; 另一方面, 在两者之间形成了p-n型异质结结构, 有利于光生电子的转移及光生电子与空穴的分离. 采用绿色环保的溴水再生法可显著恢复催化剂的光催化活性.
关键词p-n异质结; AgBr/CuO; 溴水再生法; 光催化
作为一种环境友好型技术, 光催化氧化在太阳能利用及能源开发利用方面展现出巨大的应用潜力. 自Fuiishima和Honda[1]采用TiO2作为光催化剂在紫外光照射下分解H2O制备H2以来, TiO2以其稳定性优异、价格低廉及抗光腐蚀性等特点而成为最重要的光催化材料之一. 然而, 由于TiO2的带隙较宽, 仅能吸收太阳光中的紫外线, 使其存在太阳光利用率低和光生载流子易复合等问题, 限制了其实际利用[2]. 因此, 开发并探索可高效利用太阳光的新型半导体光催化剂成为当今光催化研究领域的热点之一. 研究发现, AgBr(禁带宽度约为2.46 eV)在光照下可吸收一个光子, 产生活泼的电子与空穴, 显示出良好的可见光催化活性. 然而, AgBr在光照后易产生单质银(Ag0), 即具有光腐蚀性[3]. 通常采用将AgBr与其它光催化材料复合的方法来解决光腐蚀问题, 如将AgBr负载到TiO2[4,5], ZnO[6,7]和BiOBr[8,9]等n-型半导体材料载体上, 在一定程度上提高了AgBr的光稳定性. CuO是一种p-型过渡金属氧化物, 其禁带宽度约为1.34 eV[10], 导带位置大约在0.46 eV左右, 具有良好的光电化学性质及稳定性. 虽然CuO本身在可见光照射下不具有光催化活性. 但其与n-型半导体光催化剂复合而形成p-n异质结(例如CuO-TiO2[11,12]和CuO-ZnO[13,14])时, 则可明显提高催化剂的光催化活性.
本文采用简单且节能环保的沉淀法合成了具有p-n型异质结结构的AgBr/CuO可见光催化材料, 为了解决AgBr的光腐蚀问题, 采用溴水将催化剂表面形成的Ag0氧化为Ag+, 从而使AgBr的可见光催化活性显著恢复. 此外, 对具有p-n异质结结构的AgBr/CuO的光催化机理进行了阐述.
1实验部分
1.1试剂与仪器
氧化铜(CuO), 天津市大茂化学试剂厂; 硝酸银(AgNO3), 国药集团化学试剂有限公司; 溴化钾(KBr), 天津市化学试剂批发公司; 乙二醇(C2H6O2), 天津市天力化学试剂有限公司. 所用试剂均为分析纯, 实验用水为一次蒸馏水.
日本理学公司D/max-2500型X射线衍射(XRD)仪; 日本电子公司JSM-7001F型热场发射扫描电子显微镜(FESEM); 美国瓦里安有限公司Cary 3000型紫外-可见漫反射(UV-Vis)光谱仪; 美国赛默飞世尔科技公司ESCAL-AB 250Xi型X射线光电子能谱(XPS)分析仪.
1.2催化剂的制备
采用沉淀法制备AgBr/CuO复合催化剂. 取0.1 g CuO分散于20 mL乙二醇中, 于室温下搅拌30 min后, 将1.5 g AgNO3溶解于上述分散液中并继续搅拌30 min, 然后将1.1 g KBr加入到上述混合液中继续搅拌30 min, 经分离、蒸馏水洗涤3~4次后, 置于60 ℃鼓风干燥箱中干燥6 h, 得到灰绿色粉末催化剂AgBr/CuO(CuO与AgBr的摩尔比约为1∶6). 此外, 采用相同的步骤, 在不加CuO的条件下制备纯AgBr以作对比.
1.3光催化降解实验
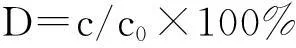
(1)式中:c0为光照前甲基橙的浓度;c为光照后甲基橙的浓度.
1.4催化剂的再生
将进行光催化降解之后的AgBr/CuO用蒸馏水洗涤后, 于60 ℃下干燥6 h, 命名为U-AgBr/CuO. 将U-AgBr/CuO复合催化剂分散于20 mL溴水溶液(质量分数3%)中, 反应30 min, 经离心、洗涤、干燥, 可得再生后的AgBr/CuO样品, 将其命名为Re-AgBr/CuO. 为了进行对比, 运用相同方法回收使用后的AgBr样品(U-AgBr)和制备再生后的AgBr样品(Re-AgBr).
2结果与讨论
2.1XRD分析
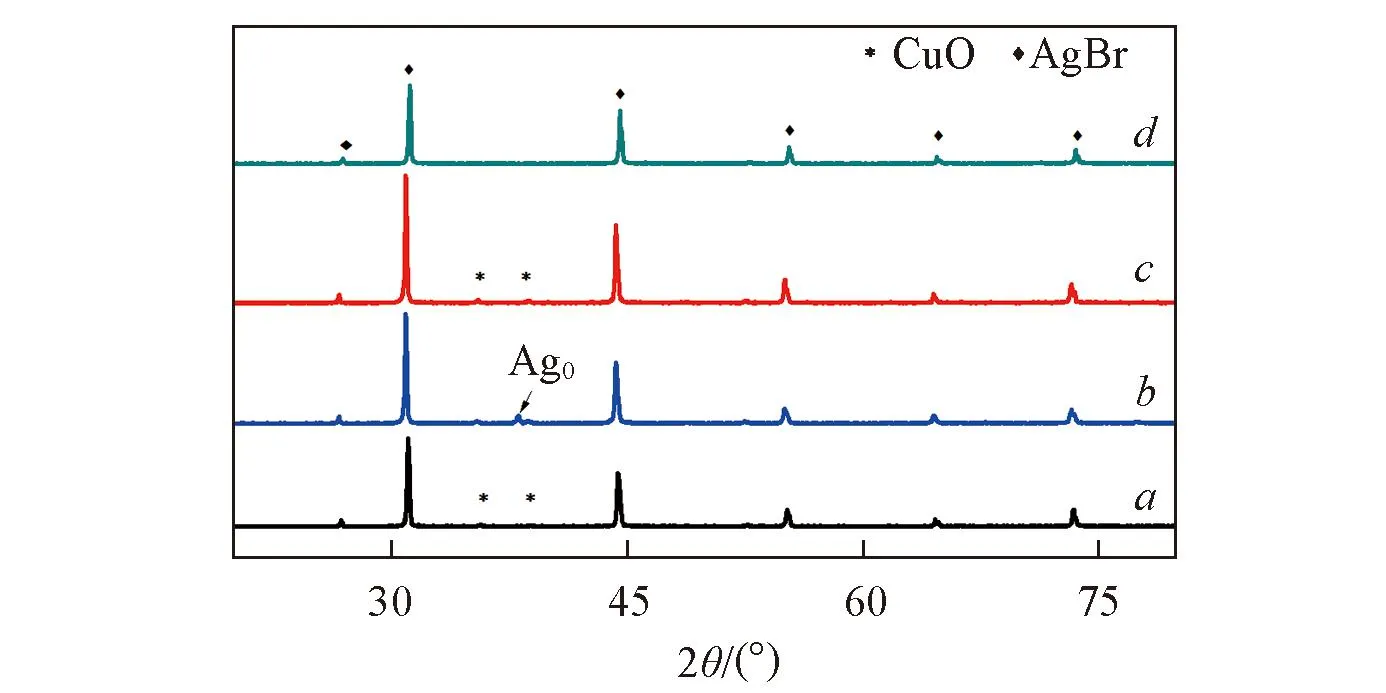
Fig.1 XRD patterns of AgBr/CuO(a), U-AgBr/CuO(b), Re-AgBr/CuO(c) and AgBr(d)
图1为AgBr/CuO, Re-AgBr/CuO和U-AgBr/CuO的XRD谱图. 图1谱线a中AgBr(JCPDS No.06-0438)[15]的衍射峰分别出现在2θ=26.78°, 30.98°, 44.39°, 52.55°, 64.52°和73.28°处. 而2θ=35.57°和38.72°处的特征峰为CuO(JCPDS No.65-2309)[16]的衍射峰. U-AgBr/CuO(如图1谱线b所示)为经过3次光降解实验后回收的催化剂, 在2θ=38.5°处出现银单质(Ag0)(JCPDS No.02-0931)的衍射峰, 这说明在光催化过程中, AgBr/CuO复合催化剂的表面发生了光腐蚀. 图1谱线c为经过溴水再生后Re-AgBr/CuO的XRD谱图, 与图1谱线b相比, 在Re-AgBr/CuO样品中单质银的特征峰消失且AgBr的衍射峰强度提高, 表明再生后催化剂的结晶度有所提高. 由此可推断, 溴水再生法可将Ag0氧化为Ag+. 根据谢乐公式[17]计算催化剂粒径:
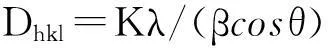
(2)
式中:K为Scherrer常数(K=0.89),D为晶粒垂直于晶面方向的平均厚度,β为实测样品衍射峰半高宽度,θ为衍射角,λ为X射线波长(0.154056 nm). 经计算, AgBr与AgBr/CuO晶体的粒径分别为82.76与64.36 nm, 表明CuO的加入抑制了AgBr的团聚.
2.2FESEM分析
图2为CuO, AgBr, AgBr/CuO和Re-AgBr/CuO的SEM照片. 由图2(B)可见, AgBr样品为平均尺寸约1.5 μm的不规则球状颗粒. 与AgBr相比, AgBr/CuO [图2(C)]颗粒尺寸在0.5~1.3 μm之间, 且催化剂的形状未发生明显改变. 而经溴水再生后的Re-AgBr/CuO [图2(D)]的颗粒尺寸比AgBr/CuO略有减小, 这表明经溴水处理后, Ag0氧化后的AgBr与CuO重组后形成粒径更小的AgBr/CuO, 且形貌未发生变化.

Fig.2 FESEM images of CuO(A), AgBr(B), AgBr/CuO(C) and Re-AgBr/CuO(D)
2.3UV-Vis吸收光谱和XPS谱分析
图3为CuO, AgBr和AgBr/CuO的紫外-可见吸收光谱及相应的(αhν)1/2-hν曲线. 由图3(A)可以看出, CuO在整个紫外及可见光区均有较强的吸收, AgBr与AgBr/CuO在485~700 nm处有明显的吸收, AgBr/CuO紫外-可见光吸收范围较AgBr略微增大. AgBr/CuO较AgBr吸收带边略有蓝移, 这是由于CuO的加入使AgBr粒径减小及两者之间的相互作用所致[18]. 这一现象与XRD结论相一致. 如图3(B)所示, 根据Kubelka-Munk公式计算[19], AgBr和AgBr/CuO的带隙能分别为1.78和1.98 eV, 这是由于CuO的复合使光催化剂的禁带宽度变大, 从而使AgBr/CuO在光照下产生光生电子与光生空穴的氧化还原能力提高.
图4为AgBr/CuO, Re-AgBr/CuO与U-AgBr/CuO的XPS谱图. 由XPS全谱图[图4(A)]可看出, 3个样品中均含有Ag, Br, O及Cu元素. 图4(B)为Ag3d的XPS谱图, 可看出AgBr/CuO中的Ag3d5/2和
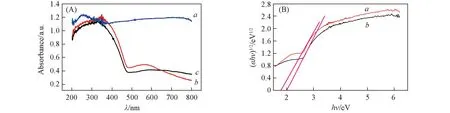
Fig.3 UV-Vis spectra for CuO(a), AgBr(b) and AgBr/CuO(c)(A) and (αhν)1/2-hν plots of AgBr(a) and AgBr/CuO(b)(B)

Fig.4 XPS spectra of AgBr/CuO(a), Re-AgBr/CuO(b) and U-AgBr/CuO(c) (A) Survey; (B) Ag3d; (C) Br3d; (D) O1s.
Ag3d3/2自旋峰分别位于367.6和373.5 eV, 而U-AgBr/CuO的Ag3d5/2和Ag3d3/2自旋峰分别位于367.9与373.9 eV, 这表明AgBr/CuO在光催化过程中发生了光腐蚀, 即在催化剂表面出现了Ag0[20,21]. 经溴水再生后, Re-AgBr/CuO中Ag3d5/2和Ag3d3/2的自旋峰位置与AgBr/CuO一致, 这表明溴水再生可将Ag0氧化为Ag+, 从而维持催化剂稳定性. 图4(C)为3个样品的Br3d的XPS分析结果. 可以看出, 3个样品的表面均存在Br-. 图4(D)为3个样品的O1s的XPS谱图. 图中位于532.3 eV的O1s自旋峰对应于催化剂表面的吸附氧[22], 且Re-AgBr/CuO的O1s峰较AgBr/CuO明显增强, 这表明在催化剂的再生过程中AgBr/CuO表面吸附了大量的氧分子.
2.4光催化性能测试
通过可见光照射下MO和苯酚溶液的降解评价了催化剂的光催化活性. 为了比较说明, 将甲基橙在不加催化剂只加光照条件下的光解过程作为空白实验. 由图5(A)可以看出, 不加催化剂时甲基橙的降解率可完全忽略不计. 而AgBr/CuO复合催化剂对MO的降解率在30 min可达到92%, 比加入纯AgBr与CuO同等条件下的降解率分别高50%与90%. 由图5(B)可见, AgBr/CuO复合催化剂对15 mg/L苯酚溶液的降解率在2 h可达到85%, 比加纯AgBr与CuO同等条件下的降解率分别高30%与80%, 这一实验结果具有与甲基橙降解类似的规律, 表明少量CuO的加入可显著提高AgBr的光催化活性.
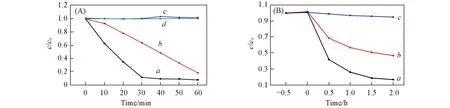
Fig.5 Degradation of MO(A) and phenol(B) under visible light irradiation with different photocatalysts a. AgBr/CuO; b. AgBr; c. CuO; d. no photocatolyst.
为进一步研究复合催化剂的稳定性和可重复利用性, 对Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr及U-AgBr的重复性进行了比较. 由图6可以看出, 经过3次重复降解实验后, Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr及U-AgBr对MO 的降解率分别为 93%, 43%, 72%和21%. 由这一结果可得出以下结论: (1) 3%溴水再生法可显著恢复AgBr/CuO与AgBr的光催化活性; (2)少量CuO的加入可提高AgBr的稳定性; (3)少量CuO的存在可加速催化剂表面的Ag0氧化为Ag+.

Fig.6 Degradation of MO with Re-AgBr/CuO, U-AgBr/CuO, Re-AgBr and U-AgBr in different cycles

Fig.7 Effects of different scavengers on the degradation of MO in the presence of AgBr/CuO and AgBr under visible-light irradiation
2.5捕获剂实验
3光催化机理
根据UV-Vis光谱分析, AgBr导带(CB)电位与价带(VB)电位位置可通过下列公式进行计算[26]:

(3)
(4)
4结论
采用沉淀法制备了n-AgBr/p-CuO异质结型可见光复合催化剂. 在模拟太阳光照射下, 0.1 g AgBr/CuO经30 min可使100 mL初始浓度15 mg/L的MO的降解率达到92%, 远远高于同等条件下AgBr与CuO对MO的降解率; AgBr与CuO复合形成的p-n异质结结构可促进光生电子-空穴的分离和光生电子的转移, 有利于光生电子与吸附在催化剂表面的O2结合产生超氧自由基, 从而提高AgBr/CuO的可见光光催化活性; 利用溴水再生法可使催化剂表面的Ag0氧化为Ag+, 迅速恢复催化剂光催化活性, 极大避免了因AgBr的光腐蚀而引起的催化剂损失.
参考文献
[1]Fujishima A., Honda K.,Nature, 1972, 238, 37—39
[2]Dahubaiyila, Wang X. H., Li X. T.,Chem.J.ChineseUniversities, 2014, 35(2), 357—361(达胡白乙拉, 王晓晖, 李晓天. 高等学校化学学报, 2014, 35(2), 357—361)
[3]Liang Y. H., Lin S. L., Liu L., Hu J. S., Cui W. Q.,Chem.J.Inorg.Chem., 2014, 30(11), 2675—2683(梁英华, 林双龙, 刘利, 胡金山, 崔文权. 无机化学学报, 2014, 30(11), 2675—2683)
[4]Hu C., Lan Y., Qu J., Hu X., Wang A.,J.Phys.Chem.B, 2006, 110, 4066—4072
[5]Wang D., Duan Y., Luo Q., Li X., An J., Bao L., Shi L.,J.Mater.Chem., 2012, 22(11), 4847—4854
[6]Krishnakumar B., Subash B., Swaminathan M.,Sep.Purif.Methods, 2012, 85, 35—44
[7]Pirhashemi M., Habibi-Yangjeh A.,Appl.Surf.Sci., 2013, 283, 1080—1088
[8]Kong L., Jiang Z., Lai H. H., Nicholls R. J., Xiao T., Jones M. O., Edwards P. P.,J.Catal., 2012, 293, 116—125
[9]Dong Y., Feng C., Zhang J., Jiang P., Wang G., Wu X., Miao H.,Chem.AsianJ., 2014, 10(3), 687—693
[10]Li P., Xu J., Jing H., Wu C., Peng H., Lu J., Yin H.,Appl.Catal.B, 2014, 156, 134—140
[11]Li G., Dimitrijevic N. M., Chen L., Rajh T., Gray K. A.,J.Phys.Chem.C, 2008, 112(48), 19040—19044
[12]Liu H., Wang Y., Liu G., Ren Y., Zhang N., Wang G., Li T.,ActaMetall., 2014, 27(1), 149—155
[13]Tseng J. C., Schmidt W., Sager U., Däuber E., Pommerin A., Weidenthaler C.,Phys.Chem.Chem.Phys., 2015, 17(18), 12282—12291
[14]Yu J., Zhuang S., Xu X., Zhu W., Feng B., Hu J.,J.Mater.Chem.A, 2015, 3(3), 1199—1207
[15]Zhang C., Ai L., Li L., Jiang J.,J.AlloysCompd., 2014, 582, 576—582
[16]Gao D., Zhang Z., Li Y., Xia B., Shi S., Xue D.,Chem.Commun., 2015, 51(6), 1151—1153
[17]Hu L. L., Du Z. P., Tai X. M., Li Q. X., Zhao Y. H.,ChineseJournalofCatalysis, 2008, 29(6), 571—576(胡利利, 杜志平, 台秀梅, 李秋小, 赵永红. 催化学报, 2008, 29(6), 571—576)
[18]Wang D., Guo L., Zhen Y., Yue L., Xue G., Fu F.,J.Mater.Chem.A, 2014, 2(30), 11716—11727
[19]Zhang X., Xie Y. J., Ma P. J., Wu Z. J., Zhao S. L., Piao L. Y.,Chem.J.ChineseUniversities, 2015, 36(10), 1977—1983(张晓, 解英娟, 马佩军, 吴志娇, 赵谡玲, 朴玲钰. 高等学校化学学报, 2015, 36(10), 1977—1983)
[20]Tian B., Wang T., Dong R., Bao S., Yang F., Zhang J.,Appl.Catal.,B:Environ., 2014, 147, 22—28
[21]Wang S., Li D., Sun C., Yang S., Guan Y., He H.,J.Mol.Catal.A:Chem., 2014, 383, 128—136
[22]Grandcolas M., Yonge L., van Overschelde O., Snyders R.,Ceram.Int., 2014, 40(8), 12939—12946
[23]Katsumata H., Hayashi T., Taniguchi M., Suzuki T., Kaneco S.,Mater.Sci.Semicond.Process, 2014, 25, 68—75
[24]Padervand M., Elahifard M. R., Meidanshahi R. V., Ghasemi S., Haghighi S., Gholami M. R.,Mater.Sci.Semicond.Process, 2012, 15(1), 73—79
[25]Wu S., Zheng H., Wu Y., Lin W., Xu T., Guan M.,Ceram.Int., 2014, 40(9), 14613—14620
[26]Cao J., Luo B., Lin H., Xu B., Chen S.,J.Hazard.Mater., 2012, 17, 107—115
Synthesis, Photocatalytic Activity and Regeneration of
AgBr/CuO Heterojunction Photocatalyst†
ZHANG Xue, LIU Jianxin, WANG Yawen, FAN Caimei, DUAN Donghong, WANG Yunfang*
(InstituteofCleanTechniqueforChemicalEngineering,TaiyuanUniversityofTechnology,Taiyuan030024,China)
AbstractAgBr/CuO p-n heterojunction photocatalyst was prepared by precipitation method and characterized by X-ray diffraction(XRD), UV-Vis spectroscopy(UV-Vis), field emission scanning electron microscopy(FESEM) and X-ray photoelectron spectroscopy(XPS), respectively. The photocatalytic mechanism was speculated through active species test and band structure analysis. Besides, the used AgBr/CuO was regenerated by 3%(mass fraction) bromine water. The results show that the degradation rate of methyl orange(c0=15 mg/L) over 0.1 g AgBr/CuO was maintained at 92% after 30 min, which was much higher than that over pure AgBr under the same conditions. The reasons for improving the photocatalytic activity of AgBr/CuO photocatalyst are that the band gap of AgBr/CuO photocatalyst become wider and the p-n heterojunction structure is formed between AgBr and CuO. The wider band gap was benefit for the redox ability of photogenerated electrons and photogenerated holes. The p-n heterojunction structure of photocatalyst could accelerate the shift of electrons and the separation of e--h+. In addition, the use of bromine water regeneration method could significantly restore photocatalytic activity.
Keywordsp-n Heterojunction; AgBr/CuO; Bromine water regeneration; Photocatalysis
(Ed.: S, Z, M)
† Supported by the National Natural Science Foundation of China(No.21176168) and the National Natural Science Foundation of Youth, China(No.21206105).
doi:10.7503/cjcu20150514
基金项目:国家自然科学基金(批准号: 21176168)和国家青年自然科学基金(批准号: 21206105)资助.
收稿日期:2015-07-06. 网络出版日期: 2015-12-20.
中图分类号O644
文献标志码A
联系人简介: 王韵芳, 女, 博士, 副教授, 主要从事光催化水处理研究. E-mail: wyfwyf53540708@sina.com
