保健食品中9种镇静催眠类药品的UPLC-MS/MS法检测及质谱裂解特征的研究
2016-03-25刘成雁王志嘉任雪冬尤海丹吴冬冬田福林
王 璐,刘成雁,王志嘉,任雪冬,尤海丹,熊 爽,吴冬冬,田福林
(辽宁省分析科学研究院,辽宁 沈阳 110015)
保健食品中9种镇静催眠类药品的UPLC-MS/MS法检测及质谱裂解特征的研究
王璐,刘成雁*,王志嘉,任雪冬,尤海丹,熊爽,吴冬冬,田福林
(辽宁省分析科学研究院,辽宁沈阳110015)
摘要:建立了同时测定保健食品中9种镇静催眠类化合物(氯美扎酮、三唑仑、阿普唑仑、艾司唑仑、地西泮、硝西泮、奥沙西泮、劳拉西泮、氯氮卓)的超高效液相色谱-串联质谱(UPLC-MS/MS)分析方法,并研究了其质谱裂解规律。样品以甲醇为提取溶剂,经Zorbax SB-C18(3.5 μm,2.1 mm×150 mm)色谱柱分离,乙腈-水(含0.1%甲酸)为流动相梯度洗脱,流速为0.3 mL/min。采用电喷雾离子源(ESI),正离子多反应监测(MRM)扫描方式检测,基质匹配标准曲线法定量。9种药物在0.5~50 μg/L范围内线性关系良好,相关系数(r2)均大于0.99,定量下限为1.6~9.2 μg·kg-1。低、中、高3个加标水平下的平均回收率为78.1%~101.2%,日内相对标准偏差(RSD)为1.4%~9.7%,日间RSD为2.0%~11.2%。该法简便、快速、准确可靠,适合于保健食品中非法添加镇静催眠类药品的高通量筛查。而对目标化合物碎片离子及质谱裂解规律的研究,也为其定性鉴别和定量分析提供了参考。
关键词:超高效液相色谱-串联质谱;保健食品;镇静催眠类;质谱裂解
失眠已成为当今社会的一种流行性疾病,安眠药为人类带来福音的同时,也给人类的健康带来了很大的问题[1-2]。但由于这些药物对失眠症的治疗见效迅速,一些厂家在生产具有催眠作用的保健食品时添加这些西药成分以提高产品的疗效,欺骗消费者,长期服用会对消费者的身体健康造成严重危害。因此有必要建立快速、准确检测保健食品中镇静催眠类药物的分析方法。
目前,国内外关于该类药品的检测方法主要有薄层色谱法(TLC)[3]、酶联免疫分析法(ELISA)[4]、高效液相色谱法(HPLC)[5-7]、气相色谱-串联质谱联用法(GC-MS/MS)[8-9]、液相色谱-串联质谱法(LC-MS/MS)[10-19]。其中TLC和HPLC法的灵敏度低,选择性和特异性较差;ELISA法的结果需借助其它方法确证;GC-MS/MS法需要衍生化,处理方法相对复杂;LC-MS/MS法具有灵敏度高、选择性好等优点,成为目前报道较多的方法。相比文献报道的LC-MS/MS[16-20]方法,本研究在目标化合物的选择上,除了检测常见的苯二氮卓类药物外,增加了国内外研究很少的氯美扎酮。采用质谱法对9种化合物的裂解规律进行了研究,首次推断了氯美扎酮的裂解方式。对9种化合物进行了完整的方法学确证,重点考察了化合物的基质效应,并采用基质加标法定量消除基质影响。该法选择性好、灵敏度高、简单快速、稳定可靠,适用于保健食品中非法添加的镇静催眠药物的定性、定量检测。
1实验部分
1.1仪器与试剂
Agilent 1200 UPLC/6410 B MS/MS 液相色谱-串联四极杆质谱联用仪(安捷伦科技有限公司);KQ3200DB型超声清洗器(江苏昆山超声仪器有限公司);乙腈、甲醇(色谱纯,美国Fisher Scientific公司);甲酸(HPLC级,美国Tedia公司);实验用水为Milli-Q纯水系统(Millipore公司)制备的超纯水;对照品:三唑仑、阿普唑仑、艾司唑仑、地西泮、硝西泮、奥沙西泮、劳拉西泮、氯氮卓、氯美扎酮均购自中国药品生物制品检定所。
1.2样品处理
准确称取0.5 g(或0.5 mL)样品于10 mL容量瓶中,加入适量甲醇,超声处理30 min,放冷,加甲醇至刻度,摇匀,过0.20 μm微孔滤膜,待测定。
1.3标准溶液配制与标准曲线
称取标准品各10.0 mg,用甲醇溶解并定容至10 mL棕色容量瓶中,配成 1.0 g/L的标准储备液,临用时以甲醇稀释成质量浓度分别为0.5,1.0,2.0,5.0,10,20,50 μg/L的系列混合标准溶液。将空白样品按“1.2”方法进行处理,得到空白基质,配制成质量浓度分别为0.5,1.0,2.0,5.0,10,20,50 μg/L的混合基质加标溶液,以各组分的峰面积对其质量浓度绘制标准曲线。
1.4测定方法
1.4.1液相色谱条件色谱柱:Zorbax-SB-C18色谱柱(2.1 mm×150 mm,3.5 μm);流动相:A为0.1%甲酸水溶液,B为乙腈,梯度洗脱程序:0~1 min,70% A;1~3 min,40%A;3~4 min,50%~40% A; 4~5 min,40%~30% A; 5~6 min,30%~70% A;6~10 min,70% A;流速为0.3 mL/min;柱温 40 ℃;进样量10 μL。
1.4.2质谱条件离子源为电喷雾(ESI)离子源,正离子电离模式;干燥气(N2)温度350 ℃,干燥气流量 9.0 L/min,雾化气(N2)压力295.4 kPa,电喷雾电压4 000 V,扫描方式为多反应监测(MRM)模式,其他质谱参数见表1。
2结果与讨论
2.1质谱条件优化及裂解规律的研究
由于9种化合物均具有仲胺或叔胺碱性基团,容易加合H+形成带正电荷的母离子[M+H]+,因此选用ESI+为离子化模式,确定分子离子峰,同时对干燥气温度、雾化气压力、干燥气流量等参数进行优化。分别对母离子进行碰撞诱导解离,选择2个信噪比较高的特征子离子分别作为定量、定性离子对,9种药物的质谱图和裂解规律如图1所示。

表1 9种镇静类药物的质谱条件参数
* quantitative ion
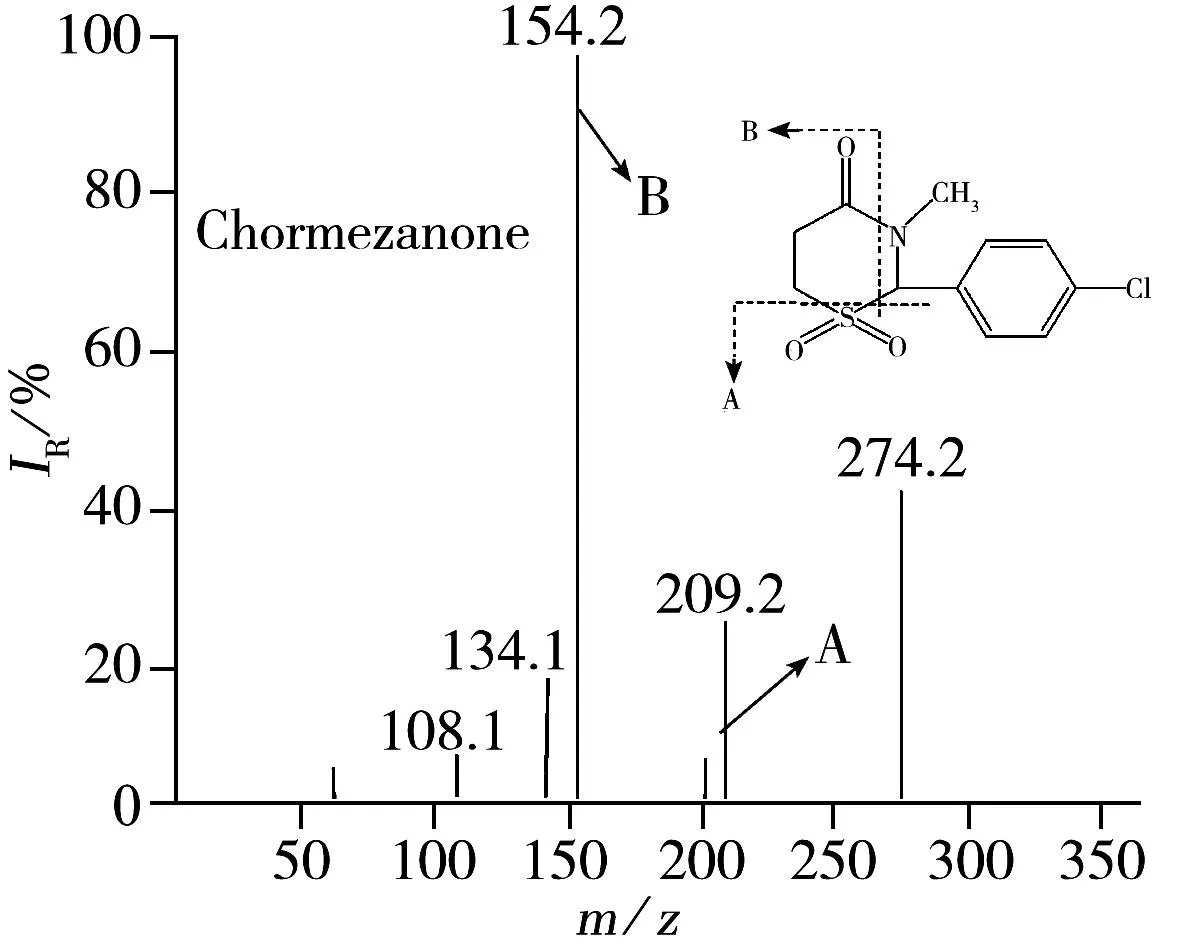
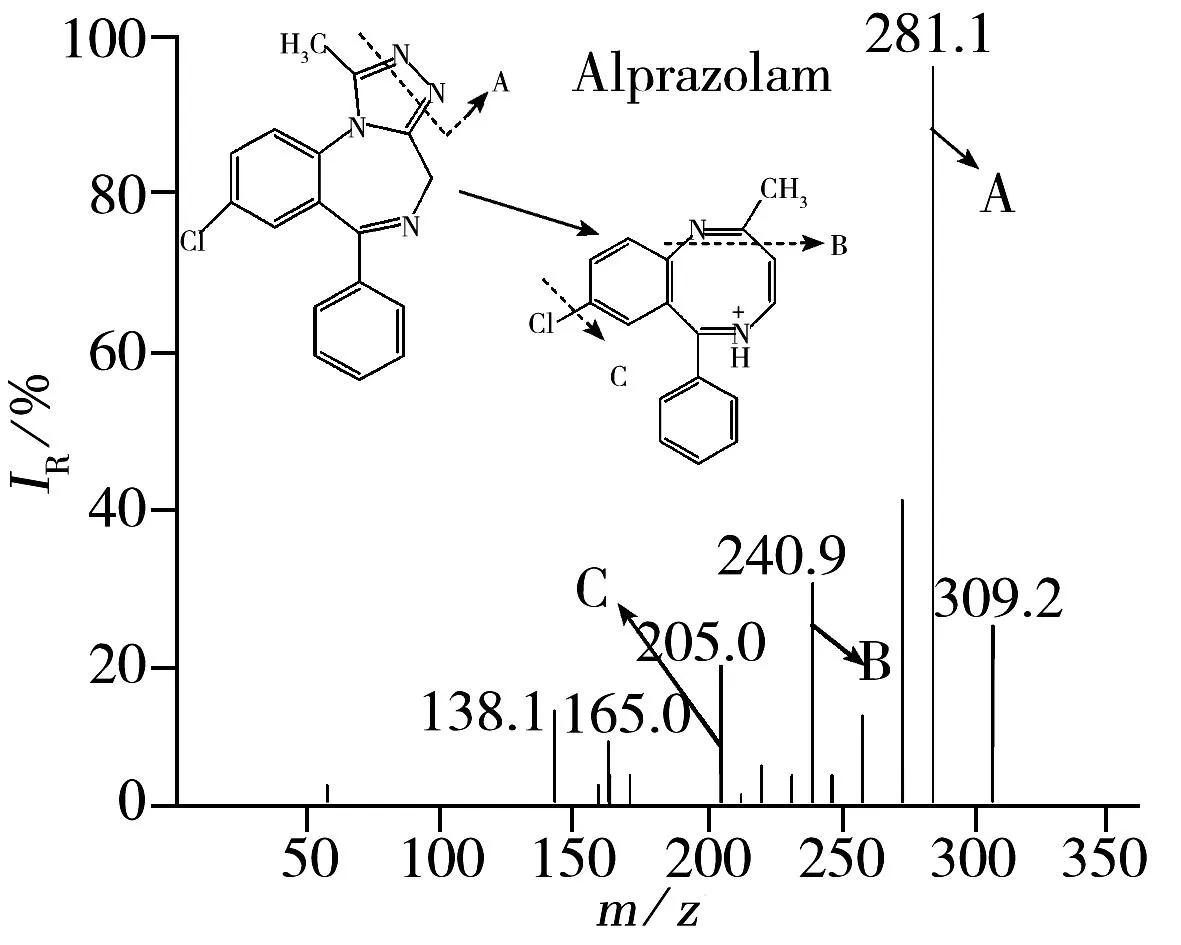

质谱数据分析结果表明:氯美扎酮磺酰基团有两个O原子,吸电子作用很强,造成C—S键不稳定,易发生断裂。结合其结构可以推断氯美扎酮的裂解机制为:磺酰基两端C—S首先发生断裂,得到m/z209的碎片离子。羰基上O原子也具有吸电子能力,造成C—N不稳定,推断氯美扎酮下一步的裂解发生在羰基上的C—N键,得到m/z154的碎片离子。三唑仑、艾司唑仑和阿普唑仑均含有三氮唑仑环,通过比较母离子与子离子之间的质量差,推断三者首先脱去的碎片离子(28)为N2分子,分别产生m/z315,267,281的碎片离子;阿普唑仑与艾司唑仑的不同之处是C(3)位连有1个甲基,所以下一步裂解分别脱去中性碎片CH3CN(41)、HCN(27),均得到m/z240的碎片离子;三唑仑同样脱去中性碎片CH3CN(41),得到m/z274的碎片离子;3个化合物最后脱去Cl原子得到m/z205的碎片离子,形成具有稳定结构的三环共轭体系。奥沙西泮和劳拉西泮首先发生C—OH键断裂,脱去H2O分子,分别得到m/z269和m/z303的碎片离子。下一步断裂发生在羰基碳上,脱去CO分子,得到m/z241和m/z275的碎片离子;地西泮脱去CO分子,得到m/z257的碎片离子;硝西泮脱去中性分子NO2,得到m/z236的碎片离子; 推测其它具体裂解方式如图1所示。电喷雾电离作为一种软电离技术,通常易丢失中性小分子,进而生成稳定性较高的离子,本研究结果也证明了这一点。
2.2液相色谱条件的优化
对比考察了有机溶剂与不同种类挥发性缓冲液作为流动相时对待测物分离效果及灵敏度的影响。通过比较发现,使用甲醇-甲酸体系时,各化合物的保留时间相对较长,而且氯美扎酮的色谱峰形较宽并有明显的拖尾现象;使用乙腈-甲酸体系时,目标化合物的出峰时间较快;使用乙腈-乙酸铵体系时,目标化合物的响应值比用乙腈-甲酸体系低。综合以上各因素,确定以乙腈-0.1%甲酸水溶液为流动相体系,梯度洗脱。9种化合物的色谱图见图2。
2.3方法专属性
取不含目标化合物的口服液、胶囊剂的混合阴性样品按“1.2”方法操作,上机测定;将一定浓度的标准混合液加入空白阴性样品中,依同法操作,所得色谱图见图2。9种目标化合物的保留时间为 2.7~10 min,而空白样品在该时间段未出峰。结果表明:空白阴性样品不干扰9种目标化合物的测定,可以用于基质匹配标准曲线法定量。

2.4基质效应的影响
目前,关于保健食品中睡眠类药物的测定研究已有文献报道[16-19],但对该类化合物基质效应的考察相对较少。本实验采用提取后添加法[20]评价基质效应:基质效应(Matrix effect,ME)=(基质匹配标准溶液峰面积对浓度所作曲线的斜率/无基质标准溶液峰面积对浓度所作曲线的斜率-1)×100%。其中,ME>0表示存在基质增强效应;ME<0表示存在基质抑制效应;ME=0表示在曲线范围浓度内基质对响应强度无影响。在0.5~50 μg/L浓度范围内,9种分析物的ME(%)值为-36.4%~22.5%(表2),氯美扎酮的基质抑制效应较大,其它化合物也存在不同程度的基质抑制或增强作用。为了补偿基质效应,采用基质加标法定量消除基质影响,以满足检测要求。
2.5线性范围与定量下限
取空白基质配制质量浓度分别为 0.5,1.0,2.0,5.0,10,20,50 μg/L的9种混合标准溶液,以各组分的峰面积(Y)对其质量浓度(X,μg/L)绘制标准曲线(见表3)。结果显示:各化合物在0.5~50 μg/L范围内呈良好的线性关系,相关系数(r2) 均大于0.99。9种化合物的检出限均能达0.5 μg/L,以信噪比为10估算9种目标物的定量下限为1.6~9.2 μg/kg。

表2 9种化合物在口服液和胶囊样品中的基质效应
a:Sm is the slope of the calibration plot with calibration solutions in solvent(Sm为无基质标准溶液峰面积对浓度所作曲线的斜率); b:Ss is the slope of calibration plot with matrix-matched calibration solutions(Ss为空白基质匹配标准溶液峰面积对浓度所作曲线的斜率)

表3 9种化合物的线性方程、线性范围、相关系数与定量下限
2.6加标回收率、准确度与精密度
以不含目标化合物的口服液和胶囊制剂作为阴性样品,分别添加1.0,5.0,20 μg/L浓度的标准溶液,进行加标回收率和精密度实验。每个浓度平行测定6次,连续测定3 d,各化合物的回收率及日内、日间相对标准偏差(RSD)如表4所示。9种化合物的平均回收率为 78.1%~101.2%,日内RSD为1.4%~9.7%,日间RSD为 2.0%~11.2%,可满足定量分析要求。
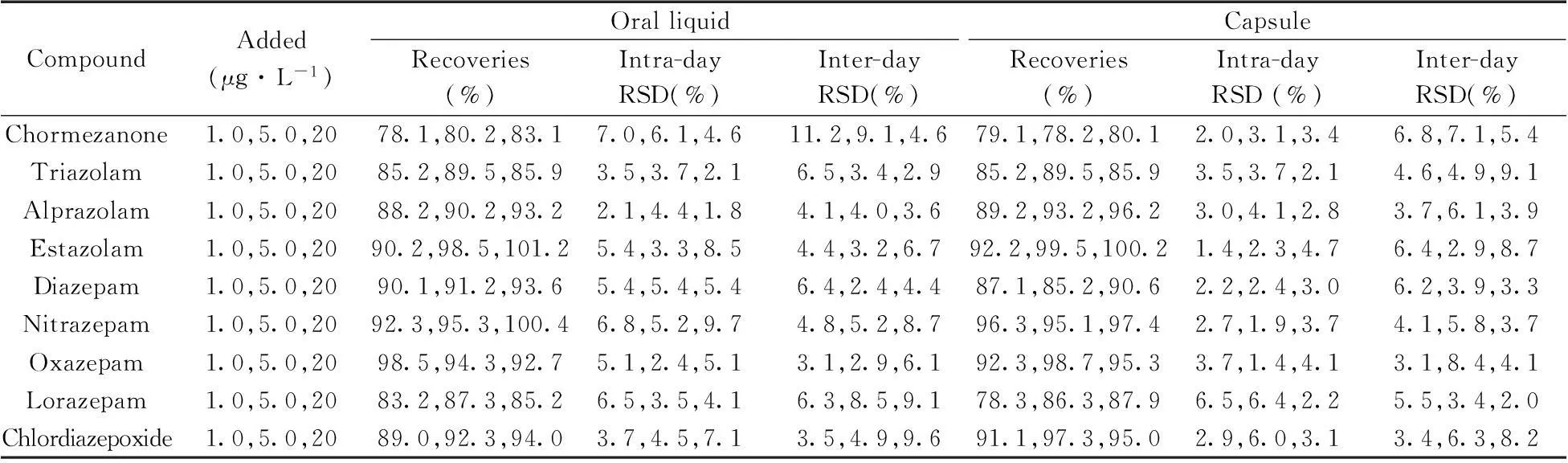
表4 9种化合物在口服液和胶囊样品中的回收率及相对标准偏差(n=6)
2.7实际样品的测定
采用上述方法检验了20批具有镇静安神催眠功效的保健食品,其中1种样品检出阿普唑仑(89.6 μg·kg-1)。
3结论
本文建立了超高效液相色谱-串联质谱法对保健食品中9种镇静催眠类药物进行检测。该法选择性好、灵敏度高、操作简便快速、稳定可靠,具有很好的实际应用价值,已应用于日常检测工作。
参考文献:
[1]Liu Y M.Chin.J.Clin.RationalDrugUse(刘玉梅.临床合理用药杂志),2012,5(23):64-65.
[2]Caplan J P,Epstein L A,Quinn D K, Stevens J R,Stern T A.Neuropsychol.Rev.,2007,17(3):363-368.
[3]Xiao L H,Liu J J,Guan X Y,Xiong Y,Wang T J,Li J.ChinaMod.Med.(肖丽和,刘吉金,关潇滢,熊英,王铁杰,李军.中国当代医药),2009,16(14):15-16.
[4]Huang W,Moody D E.J.Anal.Toxicol.,1995,19(6):333-342.
[5]He H,Sun C,Wang X R,Pham-Huy C,Chikhi-Chorfi N,Galons H,Thevenin M,Claude J R,Warnet J M.J.Chromatogr.B,2005,814(2):385-391.
[6]Capella-Peiró M E,Bose D,Martinavarro-Domínguez A,Gil-Agustí M,Esteve-Romero J.J.Chromatogr.B,2002,25(2):241-249.
[7]Qu J G,Xu Q Y.ChinaPharm.(曲建国,徐秋阳.中国药师),2010,13(12):1694-1697.
[8]Wang Z L,Zhang J L,Zhang Y N.J.Chin.MassSpectrom.Soc.(王占良,张建丽,张亦农.质谱学报),2009,30(5):282-286.
[9]Papoutsis I I,Athanaselis S A,Nikolaou P D,Pistos C M,Spiliopoulou C A,Maravelias C P.J.Pharm.Biomed.Anal.,2010,52(4):609-614.
[10]Shi Y T,Duan J,Wang H J,Zheng J,Wang J W.J.Instrum.Anal.(石银涛,段杰,王绘军,郑经,王俊伟.分析测试学报),2014,33(12):1436-1440.
[11]Toyo′oka T,Kumaki Y,Kanbori M,Kato M,Nakahara Y.J.Pharm.Biomed.Anal.,2003,30(6):1773-1778.
[12]Simonsen K W,Hermansson S,Steentoft A,Linnet K.J.Anal.Toxicol.,2010,34(6):332-341.
[13]Zhang S,Zhou S,Chen D W,Hua Z D,Wu Y N,Sun C Y,Zhao Y F.J.Instrum.Anal.(张烁,周爽,陈达炜,花镇东,吴永宁,孙承业,赵云峰.分析测试学报),2014,33(11):1213-1218.
[14]Yan L J,Zhang F,Wu M,Wu S H,Chu X G,Zhou Y.J.Instrum.Anal.(严丽娟,张峰,吴敏,吴抒怀,储晓刚,周昱.分析测试学报),2011,30(11):1301-1305.
[15]Moore C,Coulter C,Crompton K,Zumwalt M.J.Anal.Toxicol.2007,31(9):596-600.
[16]Zhu L,Ruan L P,Liu H L,Ji W L,Ma Y J.Chin.J.Chromatogr.(朱琳,阮丽萍,刘华良,吉文亮,马永建.色谱),2013,31(7):709-713.
[17]Zhang J L,Wang Z L,Zhang Y N.J.Chin.MassSpectrom.Soc.(张建丽,王占良,张亦农.质谱学报),2009,30(5):275-281.
[18]Yan A H,Li X L,Xi C X,Zhang L,Xia S,Wang G M,Tang B B,Mu Z D.Chin.J.Anal.Chem.(严爱花,李贤良,郗存显,张雷,夏爽,王国民,唐柏彬,母昭德.分析化学),2013,41(4):509-516.
[19]Pan X H.J.FoodSafetyQuality(潘小红.食品安全质量检测学报),2014,5(5):1524-1532.
[20]Matuszewski B K,Constanzer M L,Chavez-Eng C M.Anal.Chem.,1998,70(13):882-889.
Simultaneous Determination of 9 Kinds of Sedative Hypnotic Drugs in Health Foods Using UPLC-MS/MS and ESI Fragmentation Studies
WANG Lu,LIU Cheng-yan*,WANG Zhi-jia,REN Xue-dong,YOU Hai-dan,XIONG Shuang,WU Dong-dong,TIAN Fu-lin
(Liaoning Academy of Analytical Science,Shenyang110015,China)
Abstract:An ultra high performance liquid chromatography-tandem mass spectrometric(UPLC-MS/MS) method was developed for the determination of 9 sedative hypnotic drugs(chormezanone,triazolam,alprazolam,estazolam,diazepam,nitrazepam,oxazepam,lorazepam,chlordiazepoxide) in health foods, and the dissociation pathways of nine drugs were studied by electrospray ion mass spectrometry.After extracted with methanol,the analyses were separated on a Zorbax SB-C18(3.5 μm,2.1 mm×150 mm) chromatographic column by gradient elution using a mixture of acetonitrile and 0.1% formic solution as mobile phase at a flow rate of 0.3 mL/min.Detection was performed by electrospray ionization in the positive ion mode under multiple reaction monitoring(MRM) mode,and quantified with matrix standard curve.As a result,the calibration curves for all target compounds were linear in the range of 0.5-50 μg/L with correlation coefficients(r2) larger than 0.99.The limits of quantitation(LOQ,S/N≥10) were in the range of 1.6-9.2 μg·kg-1.The recoveries for all the drugs were 78.1%-101.2%.The intra- and inter-day precisions values at three spiked levels were between 1.4%-9.7%and 2.0%-11.2%,respectively.With strong selectivity,high sensitivity,easy operation and rapid detection,this method could be used to detect 9 kinds of illegally added sedative drugs in health foods.Moreover,the study of mass spectrometric behaviours and ion fragmentation patterns of nine drugs could provide a reference for the qualitative identification and quantitative analysis.
Key words:ultra high performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);functional foods;sedative hypnotic drugs;fragmentation
中图分类号:O657.63;TQ460.72
文献标识码:A 1004-4957(2016)02-0219-06
doi:10.3969/j.issn.1004-4957.2016.02.014
*通讯作者:刘成雁,博士,研究员,研究方向:危险化学品的快速分析方法和应急处理,Tel:024-24823748,E-mail:chengyanliuln@163.com
基金项目:国家自然科学基金项目(21307051);辽宁省自然科学基金项目(2013020152)
收稿日期:2015-07-29;修回日期:2015-08-24
