固相萃取/液相色谱-串联质谱法同时测定面膜类化妆品中非法添加的53种糖皮质激素
2016-03-25罗辉泰黄晓兰吴惠勤朱志鑫林晓珊马叶芬
罗辉泰,黄晓兰,吴惠勤,朱志鑫,黄 芳,林晓珊,马叶芬,
邓 欣,周培才,张秋炎,简艳婷
(中国广州分析测试中心 广东省化学危害应急检测技术重点实验室,广东 广州 510070)
约稿专栏
固相萃取/液相色谱-串联质谱法同时测定面膜类化妆品中非法添加的53种糖皮质激素
罗辉泰*,黄晓兰,吴惠勤,朱志鑫,黄芳,林晓珊,马叶芬,
邓欣,周培才,张秋炎,简艳婷
(中国广州分析测试中心广东省化学危害应急检测技术重点实验室,广东广州510070)
摘要:建立了固相萃取/液相色谱-串联质谱法(SPE/LC-MS/MS)同时测定面膜类化妆品中53种糖皮质激素的方法。样品经水分散后,加入甲醇涡旋提取,提取液用水稀释后,采用Cleanert PEP(60 mg,3 mL)固相萃取小柱净化。待测物经Waters CORTECS C18+(100 mm×2.1 mm,2.7 μm)色谱柱分离,在电喷雾离子源的正离子模式下以动态多反应监测(DMRM)模式采集数据并作定性筛查和定量分析。53种药物在相应的浓度范围内线性关系良好,相关系数均大于0.99,在3个不同浓度加标水平下,平均回收率为66.8%~106.3%,相对标准偏差(RSD)为3.9%~13.2%,检出限(LOD,S/N≥3)和定量下限(LOQ,S/N≥10)分别为3 μg/kg及10 μg/kg。该方法操作简便、定性可靠、定量准确、灵敏度高,适用于化妆品中非法添加糖皮质激素的定性确证和准确定量。
关键词:化妆品;糖皮质激素;固相萃取;液相色谱-串联质谱法;动态多反应监测;非法添加
随着电商及微商的兴起,面膜成为消费量最大的化妆品之一,但其安全性堪忧。近来,诸如“毒面膜”致消费者“毁容”的化妆品安全事件频频被各大媒体报道,因使用“激素面膜”而患上激素性皮炎的患者日益增多,引起人们的高度关注。
研究表明,糖皮质激素具有调节糖、脂肪和蛋白质生物合成和代谢的作用。临床上糖皮质激素作为抗感染药物,可抑制纤维细胞增生,减少5-羟色胺形成,因而对皮肤有一定的嫩白作用[1]。但如果长期使用含有这类物质的化妆品,将会引起皮肤变薄、毛细血管扩张、毛囊萎缩,一旦停用,皮肤就会发红、发痒,出现红斑、丘疹、脱屑等激素依赖性皮炎症状,过量使用还会引发血糖升高、高血压、骨质疏松、免疫功能下降及肥胖等危害[2]。因此,我国《化妆品卫生规范》(2007年版)[3]及欧盟化妆品规程[4]中均明确规定,化妆品中禁用糖皮质激素。
目前,针对化妆品中糖皮质激素残留的检测已有报道,方法主要为液相色谱法[5-7]及液相色谱-质谱法[8-15]。然而,这些方法可同时分析的药物种类太少、覆盖面窄,无法有效排除化妆品非法添加糖皮质激素的安全隐患。目前用于化妆品中的糖皮质激素多达数十种,而且呈增长趋势,尽管现行的国家标准方法[15]能同时检测的种类已达到41种,但尚不能满足日常监管需求,给不法商家可乘之机。本研究首次建立了同时测定面膜类化妆品中53种糖皮质激素(包括国标规定的41种以及氟轻松、地索奈德、环索奈德、卤美他松、帕拉米松、帕拉米松乙酸酯、戊酸双氟可龙、异氟泼尼松、去羟米松、卤贝他索丙酸酯、氟尼缩松及依碳氯替泼诺12种“国标外”糖皮质激素)的固相萃取/液相色谱-串联质谱法(SPE/LC-MS/MS),并实现了其中11组同分异构体的良好分离,是目前可同时分析糖皮质激素种类最多的方法。该法操作简便快速、定性可靠、定量准确,可分析的药物种类多、覆盖面广,灵敏度高,可为化妆品中糖皮质激素的定性筛查及定量分析提供技术支持,并有效地提高化妆品的安全监管、监控水平。
1实验部分
1.1仪器与试剂
Agilent 1200 SL Series RRLC/6410B Triple Quard MS快速高效液相色谱-串联四极杆质谱联用仪(美国Agilent公司);AS 3120超声波发生器(天津奥特赛恩斯仪器有限公司);Anke TDL-40B离心机(上海安亭科学仪器厂);XW-80A快速混匀器(海门市麒麟医用仪器厂)。
53种标准物质(详见表1)购自中国食品药品检定研究院、德国Dr.Ehrenstorfer公司、美国药典委员会(USP)及加拿大Toronto Research Chemicals公司。甲醇、乙腈(色谱纯,德国Merck公司);甲酸(LC-MS级,美国Sigma公司);亚铁氰化钾溶液(K4Fe(CN)6·3H2O)、乙酸锌(C4H6O4Zn·2H2O)(分析纯,广州化学试剂厂);Oasis HLB(60 mg,3 mL)固相萃取小柱(美国Waters公司);Cleanert PEP(60 mg,3 mL)固相萃取小柱(天津博纳艾杰尔科技有限公司);HyperSep Retain PEP(60 mg,3 mL)固相萃取小柱(美国Thermo Fisher公司);实验用水为二次蒸馏水。
1.2标准溶液的配制
以甲醇为溶剂,将53种糖皮质激素分别配成质量浓度为1 000 mg/L的标准储备溶液,置棕色储液瓶中于-18 ℃保存,临用时以50%乙腈稀释成适当浓度的混合标准溶液。
1.3样品预处理
1.3.1提取准确称取样品0.5 g(精确至0.01 g),置于15 mL带螺旋盖的聚丙烯离心管中,加入 2 mL 水,在快速混匀器上充分涡旋混匀1 min,加入3 mL甲醇,充分涡旋混匀2 min,移取上清液2 mL至50 mL带螺旋盖的聚丙烯离心管中,加入20 mL水以及0.2 mL亚铁氰化钾溶液(称取10.6 g亚铁氰化钾,用水溶解并定容至100 mL),涡旋混匀,再加入0.2 mL乙酸锌溶液(称取21.9 g乙酸锌,用水溶解并定容至100 mL),涡旋混匀,4 000 r/min 离心10 min,取上清液,待净化。
1.3.2净化依次用3 mL甲醇、5 mL水活化固相萃取小柱。向活化好的小柱中倒入待净化样液,保持重力自流状态,直至所有样液通过小柱,加入5 mL 10%甲醇淋洗小柱,保持自流直至液体流尽,再用洗耳球将残留在柱上的液体压出,弃去流出液。最后加入5 mL乙腈洗脱目标物,保持重力自流,待液体流尽后,用洗耳球压出残留液体,用带1 mL刻度的梨形瓶收集所有洗脱液,置于40 ℃水浴中,氮吹浓缩至近干,用50%乙腈定容至刻度,超声30 s,涡旋混匀,过0.22 μm滤膜,待上机测定。

表1 53种糖皮质激素的质谱采集参数
* quantitative ion,RT:retention time
1.4空白基质匹配混合标准溶液的配制
取阴性样品,按“1.3”方法处理,得到空白基质溶液,作为稀释剂用于配制上机测试用混合标准工作溶液。
1.5色谱与质谱条件
采用Waters CORTECS C18+(100 mm×2.1 mm,2.7 μm)色谱柱,流动相:A为0.2%甲酸水溶液,B为0.2%甲酸乙腈溶液,梯度洗脱程序:0~10.0 min,20%B;10.0~18.0 min,20%~28%B;18.0~21.0 min,28%~38%B;21.0~25.0 min,38%~40%B;25.0~28.0 min,40%~42%B;28.0~30.0 min,42%~50%B;30.0~33.0 min,50%~60%B;33.0~33.1 min,60%~90%B;33.1~35.0 min,90%B;35.0~35.1 min,90%~20%B;35.1~40.0 min,20%B。柱温:35 ℃;流速:0.4 mL/min;进样体积:5 μL。 电喷雾离子源(ESI):正离子模式;动态多反应监测(DMRM)采集方式;干燥气温度:350 ℃;干燥气流量:12.0 L/min;雾化气压力:276 kPa;毛细管电压:4 kV;MS1及MS2均为单位分辨率;53种糖皮质激素的质谱采集参数见表1。
2结果与讨论
2.1质谱与色谱条件的优化
在电喷雾离子源下,分别对浓度为1.0 μg/mL的待测物标准溶液做正离子和负离子全扫描分析,发现53种待测化合物响应最佳的准分子离子峰均在正离子模式下获得,且母离子均为[M+H]+。在此基础上,优化各化合物的碎裂电压(Fragmentor)使其母离子的响应最大化,然后对其进行子离子全扫描分析,通过优化碰撞能量(Collision energy)使其子离子的响应最大化。根据欧盟2002/657/EC决议中有关质谱分析方法必须不少于4个识别点的规定[16],为各待测药物选取质谱响应最佳的两对MRM离子对及相应的参数作为最终质谱采集参数。由于同时测定的化合物较多,采用DMRM模式进行数据采集,可在确保准确定量的情况下仍有较高的灵敏度[17]。

图1 糖皮质激素的基本结构Fig.1 Basic molecular structure of glucocorticoids
糖皮质激素为甾体类化合物,具有17个碳的环戊烷骈多氢菲基本母核结构和含有Δ4-3,20-二酮、21-羟基的功能基[18],其基本结构如图1所示。在此结构基础上,人们对C-1~2位、C-6位、C-9α位、C-11β位及C-21位等位置进行修饰而得到了种类繁多的糖皮质激素药物。因此,这些化合物的结构极为相似,本研究涉及的53种糖皮质激素中有11组同分异构体,给色谱分离条件的建立带来了较大挑战。 根据前期经验,选用高柱效、窄内径、中等长度的色谱柱Poroshell EC-C18(100 mm×2.1 mm,2.4 μm)和CORTECS C18+(100 mm×2.1 mm,2.7 μm)进行比较实验,发现后者对53种糖皮质类激素的分离效果、色谱峰形以及出峰时间均优于前者。以含0.2%甲酸的水溶液为水相,含0.2%甲酸的乙腈为有机相,优化梯度洗脱条件,经反复试验,所有待测物均可获得良好的峰形及较高的灵敏度,且所有11组同分异构体亦获得满意的分离度(如图2所示)。 53种糖皮质激素混合标准溶液的总离子流色谱图如图3所示。
2.2样品前处理条件的优化
糖皮质激素是具有甾体结构的化合物,极性较小,脂溶性好,能溶于多种有机溶剂,结合文献资料,选择甲醇、乙腈、丙酮、乙酸乙酯、乙醚5种溶剂作为提取剂进行比较实验,结果如图4所示。乙醚及乙酸乙酯等弱极性溶剂的提取率普遍较低,尤其是部分极性较大的化合物的回收率低于30%;以丙酮作提取剂时,因基质效应较大而导致回收率偏低,其原因可能是由于丙酮能溶解与待测物极性相似的杂质而增加了后续净化难度,引起基质干扰;乙腈和甲醇的提取率较理想且比较接近,基质效应相对较小,均可作为化妆品中糖皮质激素的提取剂,但考虑到乙腈的毒性更大,价格更贵,故采用甲醇为提取剂。





图3 53种糖皮质激素混合标准溶液的总离子流(TIC)色谱图Fig.3 Total ion current(TIC)chromatogram of 53 kinds of glucocorticoids mixed standard solution

图4 不同溶剂对53种糖皮质激素的提取效果Fig.4 Extraction efficiencies of 53 kinds of glucocorticoids on different solvents
化妆品是由各种具有不同功能的原料经合理调配加工而成的混合物。常用的如保湿剂(丙二醇、丁二醇等)、防腐剂(苯氧乙醇、羟苯甲酯等)、增溶剂(橄榄油、蓖麻油等)、芳香剂以及着色剂等,复杂的样品基质给净化带来了困难。对于大分子杂质,采用常用的澄清剂(亚铁氰化钾-乙酸锌溶液)进行物理絮凝,有效去除了蜡质、硬脂酸、羊毛脂等杂质[19]。
对于其他杂质,采用固相萃取净化手段。为了兼顾极性范围较大的53种化合物,选用兼具亲水亲脂性能,比传统C18填料选择范围更广的HLB填料,比较了Oasis HLB(60 mg,3 mL)、HyperSep Retain PEP(60 mg,3 mL)及Cleanert PEP(60 mg,3 mL)3款常用SPE小柱的分离效果。结果发现,3款SPE柱的回收率、重复性及净化效果均能达到测试要求,相对而言,Oasis HLB的回收率及重复性等指标更佳,但价格较高。综合考虑,选用性价比较高的Cleanert PEP小柱。在此基础上,采用浓度水平为50 μg/kg的加标样品考察了5%甲醇、10%甲醇、15%甲醇、5%乙腈、10%乙腈及15%乙腈作淋洗液时的淋洗净化效果。结果表明,10%甲醇的淋洗液可以获得理想的净化效果,且不会明显降低待测物的回收率。实验进一步考察了甲醇及乙腈两种洗脱液的回收率,对于极性较小的环索奈德,乙腈的洗脱回收率比甲醇高,且5 mL的洗脱体积能使所有待测物得到较好的回收率。
2.3基质效应的消除
在液相色谱-质谱分析中,客观存在的基质效应会对分析方法的灵敏度、精密度以及准确度造成影响,减少基质效应的方法通常包括稀释样品溶液、增加净化步骤、采用同位素内标物、配制基质匹配标准溶液以及优化色谱-质谱条件等[20-21]。基质效应通常采用基质匹配标准曲线的斜率与纯溶剂配制标准曲线的斜率之比进行评价,其比值越接近1,说明基质效应越小,反之亦然。在优化条件下,考察了样品的基质效应,结果见表2。除个别极性较大的待测物(如曲安西龙)的基质效应略大外,其余待测物的基质效应不明显,因此,本研究采用配制基质匹配标准溶液的方法,可以较好地消除基质效应带来的偏差,保证了定量结果的准确性。
2.4线性范围、回归方程与检出限
在优化条件下,对6个浓度水平的系列混合标准工作溶液进行测定。以待测物的峰面积(y)为纵坐标,对应质量浓度(x,μg/L)为横坐标作定量工作曲线,得到线性回归方程,结果见表2。53种待测物的线性范围均为2.5 ~200 μg/L,相关系数(r2)为0.997 8~0.999 8,表明各化合物具有良好的线性关系。采用标准添加法进行测定,以定量离子信噪比(S/N)大于3确定样品的检出限(LOD),S/N大于10为定量下限(LOQ),得到53种化合物的LOD均为3 μg/kg,LOQ均为10 μg/kg。
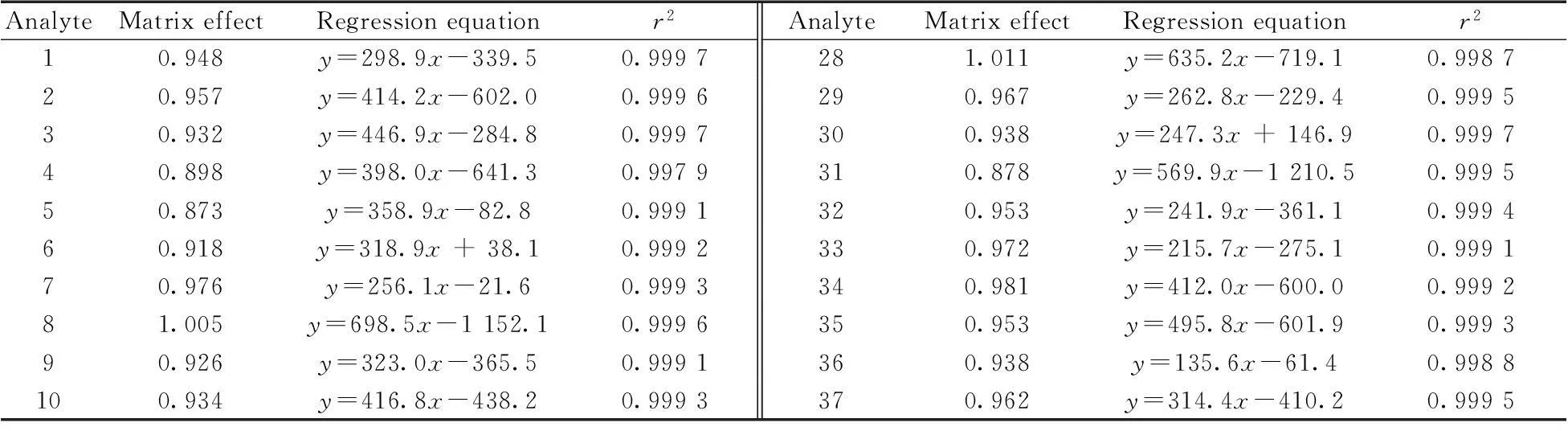
表2 53种待测物的基质效应、回归方程及相关系数
(续表2)

AnalyteMatrixeffectRegressionequationr2AnalyteMatrixeffectRegressionequationr2110.868y=429.9x+173.00.9998380.855y=271.6x-147.70.9992120.678y=326.0x-680.70.9983390.877y=437.6x-482.90.9994130.935y=258.2x-18.70.9996400.925y=341.2x-960.30.9978140.901y=332.0x-481.60.9993410.938y=211.3x-372.70.9993151.024y=394.2x+170.90.9992420.992y=186.7x-409.80.9980160.899y=198.5x-85.70.9990430.957y=471.3x-617.00.9994170.926y=297.9x-197.10.9991440.964y=291.9x-261.20.9995180.963y=185.9x-287.90.9989450.937y=309.7x-376.30.9997190.972y=297.2x-431.70.9985460.986y=298.9x-145.00.9994200.993y=333.3x-430.80.9992470.934y=372.8x-419.80.9989210.948y=340.1x-445.80.9990480.974y=311.6x-113.40.9991220.872y=229.8x-98.10.9996491.031y=481.9x-871.70.9995230.943y=508.4x+500.00.9984500.978y=522.4x-1618.80.9982240.987y=151.7x+191.90.9994510.911y=270.6x-538.40.9991250.858y=343.5x-472.80.9992520.867y=410.2x-491.80.9994260.932y=273.7x-9.00.9979530.933y=182.5x-147.30.9992270.978y=552.3x-487.80.9983
*the column number representing the compounds correspond to Table 1
2.5回收率与精密度
取阴性化妆品样品,分别进行3个浓度水平(10,50,100 μg/kg)的加标回收实验,每个加标水平均按本文的处理方法进行6个平行测定,计算得到53种待测物的平均回收率为66.8%~106.3%,相对标准偏差(RSD)为3.9%~13.2%(见表3),均符合药物残留检测要求。

表3 53种待测物的平均回收率和相对标准偏差(n=6)
*the column number representing the compounds correspond to Table 1


图5 典型阳性样品的总离子流(TIC)色谱图Fig.5 Total ion current(TIC)chromatograms of typical positive samples
2.6实际样品的测定
采用本文建立的方法,对客户送检的50款“合格”面膜(按GB/T 24800.2-2009标准方法检测,41种糖皮质激素的检测结果均为阴性样品)作53种糖皮质激素筛查分析,其中11款检出地索奈德(含量为5.6 ~106 mg/kg),5款检出氟轻松(含量为4.3~65.6 mg/kg),其余样品均未检出本文涉及的53种糖皮质激素,阳性率为32%。典型阳性样品的总离子流色谱图如图5所示。
实测结果表明,仍有不良厂商为了规避监管,添加了现行国标方法监测范围之外的糖皮质激素,以期达到速效美白祛斑的功效。同时也表明,本法能够有效、准确、可靠地测定化妆品中53种糖皮质激素,用于化妆品风险物质监测以及化妆品安全监管,可以大大提高监控水平。
参考文献:
[1]Zheng X Q,Zhou S Y,Zhou S W.CosmeticsHealthInspectionManual.Beijing:Chemical Industry Press(郑星权,周淑玉,周世伟.化妆品卫生检验手册.北京:化学工业出版社),2003:294-295.
[2]Lü G Y,Wang N P,Jin R M,Su Y M.Pharmacology.Beijing:China Press of Traditional Chinese Medicine(吕圭源,王乃平,金若敏,苏云明.药理学.北京:中国中医药出版社),2003:275-281.
[3]Ministry of Health of the People’s Republic of China.HygienicStandardforCosmetics.Beijing:China Standard Press(中华人民共和国卫生部.化妆品卫生规范.北京:中国标准出版社),2007:34.
[4]The Cosmetics Directive of the Council European Communities,Dir.76/768/EEC,March,2000.
[5]Tan J H,Xiong X T,Zhao T T,Jia F,Wang J C,Xi S F,Li H Y,Wu Y L.Mod.FoodSci.Technol.(谭建华,熊小婷,赵田甜,贾芳,王继才,席绍峰,李慧勇,吴玉銮.现代食品科技),2012,28(2):226-228.
[6]Li J,Shang S M,Chen X,Liu Y,Sun F,Song J.Chin.J.Anal.Lab.(李娟,商少明,陈新,刘瑛,孙芳,宋健.分析试验室),2013,32(5):77-80.
[7]Wu D N,Zheng H H,Wang P,Li J.Chin.J.HealthLab.Technol.(吴大南,郑和辉,王萍,李洁.中国卫生检验杂志),2008,18(2):197-198.
[8] Wang C,Ma Q,Wang X,Wu T,Bai H,Hao N,Wang J B.Chin.J.Anal.Chem.(王超,马强,王星,武婷,白桦,郝楠,王军兵.分析化学),2007,35(9):1257-1262.
[9]Chen X,Zhang X L,Liu Y,Cui H,Huang D L,Lin W X.Phys.Test.Chem.Anal.(陈溪,张晓林,刘莹,崔晗,黄大亮,林维宣.理化检验:化学分册),2013,49(4):468-472.
[10]Wang W P,Zhang M Y,Lin J,Pazilaiti Y.Chin.J.Pharm.Anal.(王伟萍,张明玥,蔺娟,帕孜来提·亚库甫.药物分析杂志),2013,33(5):837-843.
[11]Tian Y,Feng S D,Huang M H,Zhang Z J.J.ChinaPharm.Univ.(田媛,冯舒丹,黄美花,张尊建.中国药科大学学报),2011,42(1):53-57.
[12]Fiori J,Andrisanob V.J.Pharm.Biomed.Anal.,2014,91:185-192.
[13]Nama Y S,Kwon K I,Lee K B.ForensicSci.Int.,2011,210:144-148.
[14]Nama Y S,Kwon K I,Lee Y,Lee K B.ForensicSci.Int.,2012,220:e23-e28.
[15]GB/T 24800.2-2009.Determination of 41 Glucocorticoids in Cosmetics by LC-MS-MS and TLC Method.National Standards of the People’s Republic of China(化妆品中四十一种糖皮质激素的测定 液相色谱-串联质谱法和薄层色谱法.中华人民共和国国家标准).
[16] EC.Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results.Offic.J.Eur.Comm.,L221,2002:8-36.
[17]Luo H T,Huang X L,Wu H Q,Zhong Q L,Zhu Z X,Huang F,Lin X S,Xie M T,Ouyang G F.J.Instrum.Anal.(罗辉泰,黄晓兰,吴惠勤,钟巧莉,朱志鑫,黄芳,林晓珊,谢梦婷,欧阳钢锋.分析测试学报),2015,34(9):979-985.
[18]Zheng H,Weng L L,You Q D,Xu P,Xu W F,Lin H S,Xu Z,Sun T M.MedicinalChemistry.Beijing:People’s Health Publishing House(郑虎,翁玲玲,尤启冬,徐萍,徐文方,林汉森,徐正,孙铁民.药物化学.北京:人民卫生出版社),2006:376.
[19]Pan W,Li P.ChinaScienceandTechnologyAchievements(潘炜,李鹏.中国科技成果),2011,12(7):47-48.
[20]Van Eeckhaut A,Lanckmans K,Sarre S,Smolders I,Michotte Y.J.Chromatogr.B,2009,877:2198-2207.
[21]Xiang P,Shen M,Zhuo X Y.J.Instrum.Anal.(向平,沈敏,卓先义.分析测试学报),2009,28(6):753-756.
Simultaneous Determination of 53 Kinds of Glucocorticoids Illegally Added in Facial Mask Cosmetics by Solid-phase Extraction with Liquid Chromatography-Tandem Mass Spectrometry
LUO Hui-tai*,HUANG Xiao-lan,WU Hui-qin,ZHU Zhi-xin,HUANG Fang,LIN Xiao-shan,MA Ye-fen,DENG Xin,ZHOU Pei-cai,ZHANG Qiu-yan,JIAN Yan-ting
(Guangdong Provincial Key Laboratory of Emergency Test for Dangerous Chemicals,China National Analytical Center (Guangzhou),Guangzhou510070,China)
Abstract:A comprehensive analytical method was developed and validated for the simultaneous determination of 53 kinds of glucocorticoids in facial mask cosmetics using solid-phase extraction(SPE)coupled with liquid chromatography-tandem mass spectrometry(LC-MS/MS).Cosmetic sample was dispersed with water,and the analytes were extracted with methanol by vortex mixer.The extracts were cleaned up with a Cleanert PEP(60 mg,3 mL) solid phase extraction cartridge after diluted with water.Electrospray ionization mass spectrometry was performed in the positive mode using dynamic multiple reaction monitor(DMRM) mode for the qualitative and quantitative analyses of 53 kinds of analytes after separation on a column of Waters CORTECS C18+(100 mm×2.1 mm,2.7 μm).The correlation coefficients for linear calibration curves were over 0.99 in the corresponding concentration range.The average recoveries of 53 drugs from spiked sample at three different concentration levels ranged from 66.8%to 106.3%, with relative standard deviation(RSDs)of 3.9%-13.2%.The limits of detection(LODs,S/N≥3)and quantitation(LOQs,S/N≥10)were 3 μg/kg and 10 μg/kg,respectively.The established method is simple,reliable,accurate and sensitive,and is suitable for the determination of glucocorticoids illegally added in cosmetics.
Key words:cosmetics;glucocorticoids;solid-phase extraction(SPE);liquid chromatography-tandem mass spectrometry(LC-MS/MS);dynamic multiple reaction monitoring(DMRM);illegal addition
中图分类号:O657.63;O629.8
文献标识码:A
文章编号:1004-4957(2016)02-0119-08
doi:10.3969/j.issn.1004-4957.2016.02.001
*通讯作者:罗辉泰,助理研究员,研究方向:色谱-质谱分析技术,Tel:020-37656312,E-mail:luohuitai@qq.com
基金项目:广东省科技计划项目“公益研究与能力建设”专项(2014A040401038)
收稿日期:2015-10-07;修回日期:2015-12-01
