丝状真菌里氏木霉中shRNA沉默红色荧光基因
2016-02-13李洁璇刘雯莉梁智成王绍文梁秀怡佘炜怡刘刚田生礼
李洁璇,刘雯莉,梁智成,王绍文,梁秀怡,佘炜怡,刘刚,田生礼*
(1.深圳大学生命科学与海洋学院深圳市微生物基因工程重点实验室,广东深圳518060; 2.深圳市海洋生态与生物资源重点实验室深圳大学,广东深圳518060)
丝状真菌里氏木霉中shRNA沉默红色荧光基因
李洁璇1,刘雯莉1,梁智成2,王绍文2,梁秀怡1,佘炜怡1,刘刚1,田生礼1*
(1.深圳大学生命科学与海洋学院深圳市微生物基因工程重点实验室,广东深圳518060; 2.深圳市海洋生态与生物资源重点实验室深圳大学,广东深圳518060)
报道了在里氏木霉中建立的一种以红色荧光蛋白(DsRed)为报告基因的RNA干扰方法。首先,将构建的表达DsRed质粒pANRed1转化里氏木霉QM9414,得到抗潮霉素B抗性并能稳定表达DsRed的菌株DsRed-T.reesei。其次,以丙酮酸脱氢酶启动子Ppdc和纤维二糖水解酶I终止子cbh I为原件,克隆到载体pPHL上构建质粒pPHL-Ppdc-Tcbh1。根据DsRed基因序列设计特定的siRNA干扰序列和另一条无同源序列的siRNA作为阴性对照,克隆到载体pPHL-Ppdc-Tcbh1得到重组质粒。将其转化到DsRed-T.reesei中,用含有100 μg/mL潮霉素B和250 μg/mL腐草霉素的PDA平板筛选转化子。结果表明,约79%的转化子出现红色荧光沉默现象,其中一些转化子DsRed的表达几乎完全被抑制。荧光定量PCR和Western印迹分析显示DsRed基因的表达受到不同程度的下调。以上结果提示,在里氏木霉中可用此方法研究基因表达调控。
RNA干扰;shRNA;红色荧光蛋白DsRed;里氏木霉
KeywordsRNA interference;shRNA;DsRed;Trichoderma reesei
里氏木霉是一种丝状真菌,广泛应用于水解酶的生产[1],在生物降解中具有十分重要的作用。里氏木霉主要生产纤维素酶和纤维二糖水解酶I (CBHI),能合成大约64%~80%的胞外蛋白[2]。里氏木霉除了具有优越的蛋白分泌能力外,还能对蛋白进行糖基化修饰和翻译后修饰。因此,里氏木霉是重组蛋白表达和生产的优良宿主。先前通过诱导突变使得里氏木霉的纤维素酶产量显著提高,但通过此方法很难进一步提高产量。随着里氏木霉基因组测序结果的发布,使通过基因工程方法改良生产纤维素酶菌株成为可能[3]。因此,引入一种高效的基因操作手段来研究里氏木霉中纤维素酶和半纤维素基因的调控机制是可行的。在细胞、酵母和真菌中,RNA干扰是一种高效的基因沉默手段[4-7]。以基因家族中的保守序列为靶目标,RNA干扰可应用于沉默同源基因,或者从不同基因获得的嵌合序列为靶目标沉默非同源基因[8]。以绿色荧光蛋白GFP或红色荧光蛋白DsRed为报告基因的RNA干扰体系已在丝状真菌中发展起来[9-10]。在真菌中发现的第一种siRNA是用于沉默与内源基因具有同源性的外源序列[6]。据报道,合成外源的dsRNAs或者内部转录合成的dsRNAs在真菌内都能有效的沉默基因[8]。另外,dsRNAs由可诱导的启动子起始转录,能在特定的阶段研究基因表达情况[11]。丝状真菌通常含有多细胞、多核菌丝,使基因打靶的效率低。因此,RNA干扰为丝状真菌提供了一种可用于基因敲除的通用灵活手段,其中包括里氏木霉[12-13]。为了研究真菌基因的功能或提高工业菌株的特性,研究者利用各种基因操作方法在真菌体内通过同源重组破坏靶基因的完整[1,14]。然而,在许多类型的真菌中,由于转化DNA的异位整合导致[15-16]同源重组的效率通常很低,大约在0~40%不等。所以,在丝状真菌中单独敲除一个靶基因是一项艰难的工程,引入一些基因操作手段如RNA干扰是十分必要的。我们已在丝状真菌康氏木霉YC01中成功建立RNA干扰体系。体系主要由一个表达盒组成,包括里氏木霉甘油醛-3-磷酸脱氢酶启动子gpd和纤维二糖水解酶I终止子cbh1,还有一个带有内含子的发夹RNA(ihpRNA)[9]。其中包含2个430 bp的DsRed片段和一个209 bp的间隔片段,间隔片段来源于里氏木霉egl2基因的内含子2。这3个片段通过overlap PCR相连,接着插入到载体pPHL上得到重组质粒pPHL-Redi。本研究以DsRed为报告基因,获得能有效应用于工业菌株丝状里氏木霉QM9414的siRNAs,以期在DsRed-T.reesei菌株中以DsRed特定序列为靶目标的19ntsiRNAs能有效沉默DsRed的表达。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒大肠埃希菌DH5α(supE44 ΔlacU169(F80 lacZ ΔM15)hsdR17 recA1 endA1 gyrA96 thi-1 relA1)用于构建质粒,大肠埃希菌Top10F'(Invitrogen)用于载体构建和保存。质粒pANRed1从pAN7-1(GenBankaccession,Z32698)改造得到[9]。质粒pUC-19(GenBank accession,M77789)用于构建重组质粒pPHL。将质粒pAN-Red1转入里氏木霉QM9414(ATCC 26921)得到重组菌株DsRed-T.reesei。
1.1.2 培养基LB培养基:胰蛋白胨10 g/L,酵母提取物5 g/L,NaCl 10 g/L,用于平板培养时加入15 g/L琼脂,用于E.coli转化时加入100 μg/ mL氨苄青霉素(Invitrogen,美国,加利福尼亚)。PDA培养基:取马铃薯200 g,去皮切碎后加至约1 L蒸馏水中,煮沸30 min,用4层纱布过滤,收集滤液。加入20 g葡萄糖和18 g琼脂后定容至1 L,灭菌倒平板。进行T.reesei转化子筛选时加入100 μg/mL潮霉素。基本培养基:KH2PO42 g/L,(NH4)2SO41.4 g/L,尿素0.3 g/L,CaCl2·2H2O 0.4 g/L,FeSO4·7H2O 0.005 g/L,MnSO4·H2O 0.001 6 g/L,ZnSO4·7H2O 0.001 7 g/L,CoCl2· 6H2O 0.003 7 g/L,蛋白胨2 g/L,吐温80 1 g/L以及葡萄糖20 g/L。
1.1.3 试剂限制性内切酶、DNase I购于Fermentas公司,哈茨木霉溶壁酶购于Sigma-Aldrich公司,RNA提取试剂盒购于BioTeke公司,反转录试剂盒和SYBR Premix Ex Taq购于Takara公司,DsRed单克隆抗体、羊抗鼠IgG-HRP和Anti-alphaTubulin购于艾美捷公司,Bradford试剂及其他试剂购于生工生物工程公司,其他试剂均属于分析纯。1.1.4仪器高压自动灭菌锅(Hirayama,HVE-50);超净工作台(苏州净化设备总厂,SW-CJID);荧光定量PCR仪(Applied Biosystem,ViiATM7);PCR仪(ABI,2720);冷冻离心机(Thermo Scientific,Multifuge XIR);小型离心机(Eppendorf,Minispin);pH计(Mettler-Toledo,320-S);电泳仪(Amersham Pharmacia,EPS-601);水平电泳槽(北京六一,DYCP-31D);高纯水仪(力新仪器有限公司,Heal force ROP3);核酸成像系统(Bio-Rad,GEL DOX XR+);低温摇床(上海苏坤,SKY-200B);低温培养箱(上海爱朗,LTI-700);奥林匹斯BX51TRF System(东京,日本)。
1.2 方法
1.2.1 RNAi载体的构建使用引物PHL-F和PHL-R(见表1),利用pPHL-Redi载体为模板,通过PCR扩增出腐草霉素抗性基因表达盒PHL (2 784 bp),同时引入酶切位点Hind III和Pst I。经过双酶切后,插入经相同酶切的载体pUC-19中得到重组质粒pPHL[9]。以里氏木霉基因组DNA为模板,Ppdc-F和Ppdc-R(表1)为引物PCR扩增出丙酮酸脱羧化酶启动子Ppdc(1 534 bp),经Pst I和Sal I双酶切后插入到质粒pPHL得到重组质粒pPHL-Ppdc。以里氏木霉基因组DNA为模板,Tcbh1-F和Tcbh1-R(表1)为引物PCR扩增纤维二糖水解酶I终止子Tcbh1(702 bp),经BamH I和EcoR I双酶切后插入到质粒pPHL-Ppdc得到重组质粒pPHL-Ppdc-Tcbh1。将基因DsRed的mRNA输入到Applied Biosystems Website设计siRNA靶序列,选择其中两条干扰片段命名为Red-T3和Red-T12,另选一条作为阴性对照,命名为DsRed-Neg。59-bp的siRNA干扰片段包含9-nt茎环结构,连接19-nt DsRed不同靶位点正向单链siRNA和19-nt反向单链siRNA,两端各添加酶切位点Sal I和BamH I黏性末端对应的突出序列,由Life technologies公司合成siRNA寡核苷酸链(见表2)。成对寡核苷酸链利用PCR仪进行退火,反应程序:95℃加热2 min,设置PCR仪使每秒下降0.1℃,降至25℃(约90 min),结束后保存在4℃。这3个退火产物插入到经相同双酶切后的载体pPHL-Ppdc-Tcbh1中,分别得到干扰载体pPHL-Ppdc-T3-Tcbh1、pPHL-Ppdc-T12-Tcbh1和pPHL-Ppdc-Neg-Tcbh1。

表1 构建重组质粒pPHL-Ppdc-Tcbh1使用的引物Table 1Primers amplified for phleomycin gene,Ppdc and Tcbh1

表2 基因DsRed的siRNA干扰片段Table 2Sequences of siRNA targeting on DsRed gene
1.2.2 真菌转化和转化子分析参考Penttil¨a等[17]的实验方法,用聚乙二醇进行原生质体的制备与里氏木霉转化。哈茨木霉的溶壁酶融于1 mol/mL MgSO4用于制备原生质体。转化时,使用20 μg质粒DNA(pANRed1或pPpdc-shRNAs-Tcbh1)和200 μL PEG buffer。包含100 μg/mL潮霉素B(用于pAN-Red1)或100 μg/mL潮霉素B和250 μg/mL腐草霉素(用于pPpdc-shRNAs-Tcbh1)的PDA固体平板用于筛选转化子,生长2~3 d。用牙签挑取转化子接种到新的筛选培养基上,并连续筛选培养观察6代。提取转化子的DNA经PCR鉴定确认shRNA表达盒子插入到转化子的基因组中。
1.2.3 基因组DNA提取真空抽滤获取液体培养的转化子菌丝,用液氮研磨成干粉后冻于-80℃。参考Seiboth等[18]的方法提取基因组。利用RNA提取试剂盒按照说明书的方法从冷冻菌丝中提取总RNA。用DNase I处理RNA样品以去除基因组DNA。用GeneQuant 1300分光光度计和琼脂糖凝胶电泳检测RNA样品的浓度和质量。
1.2.4 RNA反转录和荧光定量PCR使用反转录试剂盒RT-PCR kit,以oligo(dT)和随机六聚体当引物,将约500 ng RNA反转录成cDNA。使用仪器进行荧光定量PCR。每个反应20 μL体积,包括2 μL模板(160稀释反转录产物),10 μL 2×SYBR Premix Ex Taq,300 nmol/L正反向引物(见表1)和nuclease-free水。程序设置:预变性95℃30 s,95℃变性5 s和60℃退火延伸31 s,40个循环。结束后得到融解曲线,用以核对PCR产物的特异性。所有PCR反应每个样品做3个复孔。结合ABI公司的软件进行分析得到相应的标准曲线。基因DsRed的表达量通过内参基因肌动蛋白校正,以对照菌株的表达量为1,其他转化子的表达量与对照样品的比值作为数据分析。
1.2.5 DsRed荧光成像与测量使用奥林匹斯BX51TRF System观察重组菌株的红色荧光。红色荧光在554 nm下被激发,用U-25ND6滤光器可检测到红色荧光。拍下荧光图像,用软件Image-Pro Plus 5.0分析红色荧光的强弱。
1.2.6 SDS-PAGE电泳检测和蛋白质印迹分析将菌株QM9414,DsRed-T.reesei菌株和干扰重组菌分别接种于PDA平板上,于28℃恒温培养7 d后,用无菌水制备孢子悬液,取大约105个孢子接种到液体培养基上,28℃、250 r/min震荡培养72 h。菌液真空抽滤后用液氮快速研磨成干粉,取约1 g干粉加入1 mL lysis buffer震荡混匀3 min后4℃冰浴30 min,4℃、12 000 r/min离心5 min。吸取上清液转移到新的1.5 mL EP管中,得到蛋白样品。使用Bradford试剂,以牛血清蛋白(BSA)为标准品,测定蛋白样品的浓度。每个蛋白样品取60 mg进行SDS-PAGE电泳并转膜到硝酸纤维素酶上。根据说明书使用DsRed单克隆抗体作为一抗进行免疫印迹,二抗使用羊抗鼠IgG-HRP,内参抗体使用Anti-alpha Tubulin。
1.2.7 数据分析每个样品的检测都做了3个复孔。使用单向方差分析实验结果,从3个独立的结果得到标准差(SD),结果均表示为(均值±SD)。在所有的结果中,P<0.05表示差异显著。
2 结果与分析
2.1 筛选稳定表达DsRed的重组菌株DsRed-T.reesei
本研究构建质粒pAN-Red1并转化到T.reesei QM9414中,含有100 μg/mL潮霉素B的PDA固体平板筛选转化子。为了获得单克隆转化子,洗脱孢子得到孢子悬液后接种到筛选平板上,连续接种培养至少8次,最后获得重组菌株DsRed-T.reesei,并将其作为研究siRNA沉默的宿主菌。在荧光显微下可观察此菌株稳定表达强红色荧光蛋白(图1)。
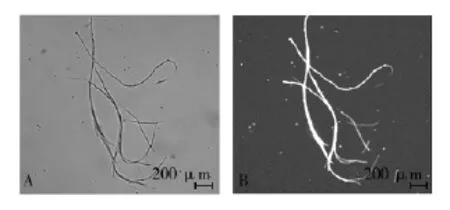
图1 质粒pAN-Red1转入T.reesei QM9414获得表达DsRed的重组菌株DsRed-T.reeseiFig.1T.reesei QM9414 transformed by plasmid pAN-Red1 generating recombinant DsRed-T.reesei strain重组菌株DsRed-T.reesei的菌丝分别在普通光源(A)和荧光光源(B)下的影像,(普通光源曝光时间:2 msec;荧光光源曝光时间:50 msecThe images of the mycelia of recombinant strain DsRed-T.reesei under ordinary light(A)and fluorescent light(B) respectively.Bars indicate 200 μm(exposed time under ordinary light:2 msec;exposed time under fluorescent light:50 msec)
2.2 RNAi下调DsRed的表达
本研究构建了重组质粒pPHL-Ppdc-T3-Tcbh1、pPHL-Ppdc-T12-Tcbh1和pPHL-Ppdc-Neg-Tcbh1,分别与质粒pAN7-1共转入DsRed-T.reesei中得到38株里氏木霉干扰转化子,具有潮霉素B和腐草霉素抗性。其中,23株T.reesei-DsRed-T3转化子,15株T.reesei-DsRed-T12转化子,另外作为对照获得17株T.reesei-DsRed-Neg转化子。将干扰转化子接种到基本培养基培养3 d后用荧光显微镜观察红色荧光的情况。根据转化子与对照菌株的荧光强度相比,将重组菌株分为3个等级,每个等级的个数占全部转化子的比例如表3所示。数据显示,干扰载体T.reesei-DsRed-T3的转化子中干扰效果显著的大约占87%,这是沉默效果最好的。T.reesei-DsRed-T12的转化子中干扰效果显著的大约为64%。然而,在DsRed-T3和DsRed-T12的转化子中,29.4%和33.3%的抗性转化子都显示红色荧光不同程度的减弱。荧光显微镜的观察结果见图2。图像结果显示,本研究构建的质粒pPHL-Ppdc-shRNA-Tcbh1中表达的shRNAs确实干扰和沉默了基因DsRed的表达,其中转化子DsRed-T3-11、DsRed-T3-19、DsRed-T12-4和DsRed-T12-8的干扰效果较好。

表3 DsRed干扰转化子的干扰效率Table 3Silenced DsRed Transformants of DsRed-T.reesei after transformation with shRNAs

图2 重组菌株DsRed-T.reesei中的干扰效果Fig.2Silencing efficiency of DsRed by siRNAs in recombinant DsRed-T.reeseiW:第1列和第3列的菌丝影像为普通光源下拍摄;R:第2列和第4列的菌丝影像为荧光光源下拍摄W:First and third columns indicate image of the mycelia in bright field;R:Second and fourth columns indicate image of the mycelia in DsRed fluorescence
2.3 里氏木霉中DsRed开放阅读框和shRNA干扰表达盒的完整性检测
上述结果初步证明shRNA在T.reesei中能有效沉默DsRed。为排除荧光强度减弱是由里氏木霉的基因组损伤或DsRed表达盒不完整所致的可能性,需要进一步实验确认,质粒pANRed1和pPHL-Ppdc-shRNAs-Tcbh1转化DsRed-T.reesei经同源重组后,DsRed基因和siRNA表达盒的完整性。分别提取DsRed-T3-11、DsRed-T3-19、DsRed-T12-4、DsRed-T12-8和阴性对照DsRed-Neg-12与DsRed-Neg-24的基因组DNA作为模板,用引物(见表4)PCR分别扩增出DsRed开放阅读框和shRNA干扰表达盒。结果显示,6个样品均能扩增出678 bp的DsRed编码序列(图3A)和754 bp的干扰表达盒子(图3B)。

表4 用于检测DsRed开放阅读框和shRNA干扰表达盒的完整性的引物Table 4Primers used in analysis of the integrity of DsRed ORF and shRNA expression cassette

图3 凝胶电泳分析重组菌的DsRed开放阅读框和shRNA干扰表达盒Fig.3Electrophorestic analysis of DsRed ORF and shRNA expression cassette for recombinantA:DsRed开放阅读框的PCR产物电泳;B:shRNA干扰表达盒的PCR产物电泳;M:DL2 000 bp DNA Marker;1~6:分别是重组菌株DsRed-T3-11、DsRed-T3-19、DsRed-T12-4、DsRed-T12-8、DsRed-Neg-12和DsRed-Neg-24A:Electrophoresis of amplified fragments of DsRed ORF;B:Electrophoresis of amplified fragments of the shRNA expression cassette;M:DL2 000 bp DNA Marker;1-6:The recombinant strains DsRed-T3-11,DsRed-T3-19,DsRed-T12-4,DsRed-T12-8,DsRed-Neg-12,DsRed-Neg-24
综合上述结果表明,转化后DsRed开放阅读框和shRNA干扰表达盒完整,确认转化子荧光强度的减弱来源于siRNA对靶基因DsRed的干扰,排除DsRed表达盒不完整或排除机体的衰减而导致荧光减弱的可能性。此外,还证明了siRNA干扰片段对里氏木霉DsRed的干扰作用发生在转录及转录后水平。
2.4 荧光定量PCR分析DsRed基因的表达量
为了进一步确认干扰重组菌中DsRed基因的下调情况,本研究进行QPCR检测DsRed基因的mRNA表达水平。结果显示,与原始菌株DsRed-T.ressei相比,干扰重组菌DsRed-T3-11、 DsRed-T3-19、DsRed-T12-4和DsRed-T12-8的DsRed基因表达水平都有不同程度的下降,分别是67.83%、99.89%、99.8%和100%。另外两个干扰重组菌DsRed-Neg-12和DsRed-Neg-24并未显示DsRed基因表达水平的下降(图4)。
另外,为探究重组菌干扰效果的时间效应,选择含有siRNA干扰片段T3的2个重组菌DsRed-T3-11和DsRed-T3-19,并以出发菌株DsRed-T.ressei为对照,进行QPCR检测。如图5所示,重组菌DsRed-T3-11在培养48、72和96 h时的DsRed相对表达量分别是各时段出发菌株DsRed-T.ressei表达量的33%、30%和32%,重组菌DsRed-T3-11的表达量则分别是出发菌株的0.2%、0%和0.01%。结果表明,同一株干扰重组菌在相同培养基中培养48、72和96 h时能均保持几乎相同的干扰效果,说明干扰效果十分稳定。

图4 重组菌在液体培养基中培养48 h后的DsRed基因相对表达量Fig.4Q-RT-PCR detection of mRNA expression level of DsRed in recombinant after incubation for 48 h出发菌株的DsRed基因相对表达量,设为1。误差棒代表标准差(珚X±SD,n=3,*P<0.05,**P<0.01),图5同Error bars represent standard deviations(珚X±SD,n=3,*P<0.05,**P<0.01),same figure 5
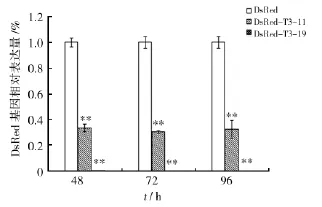
图5 重组菌DsRed-T3-11和DsRed-T3-19培养48、72和96 h的DsRed基因相对表达量Fig.5Q-RT-PCR detection of mRNA expression level of DsRed gene in tansformants after incubation for 48,72 and 96 h respectively
2.5 干扰重组菌的蛋白质印迹分析
为了确认干扰重组菌中mRNA表达水平和蛋白合成水平一致,本研究进行了蛋白质印迹。从原始菌株T.reesei QM9414、菌株DsRed-T.reesei、干扰重组菌DsRed-neg-12和DsRed-T3-19中提取蛋白样品。结果如图6所示,菌株DsRed-T.reesei和DsRed-neg-12样品显示了明显的蛋白条带,而菌株T.reesei 9414和干扰重组菌DsRed-T3-19并未显示蛋白条带。综合这些结果表明,蛋白质印迹结果与QPCR检测结果完全一致。

图6 重组菌DsRed-T3-19和对照组的蛋白质印迹分析Fig.6Western bolt assay of transformant s and controls for 72 h respectivelyT.reesei QM9414:原始菌株;DsRed-T.reesei:能稳定表达DsRed的重组菌株;DsRed-neg-12:转入阴性对照片段的重组菌株;DsRed-T3-19:转入有效干扰片段的重组菌株;以上菌株均在液体培养72 hT.reesei QM9414:Host cell;DsRed-T.reesei:The transformant with expression of DsRed;DsRed-neg-12:The transformant with negative sIRNA;DsRed-T3-19:The transformant with effective sIRNA;Both strains are cultured for 72 h
3 讨论
RNA干扰体系已在不同的生物中建立发展起来,多用绿色荧光蛋白(GFP)作为报告基因,便于使用荧光显微镜从平板上筛选转化子[19-20]。GFP已成功用于各种微生物中[21-23],宿主细胞携带一个gfp基因并在荧光显微镜下发出绿色荧光。然而,原始菌株康氏木霉在GFP的波长上显示出一定程度的内源性荧光,在筛选过程中会出现假阳性转化子[9]。在子囊真菌如覃青霉、绿木霉和哈茨木霉中,红色荧光蛋白已成功表达,并在顶头孢霉中作为报告基因进行RNA干扰[9,24-25]。但与里氏木霉相关的研究从未报道。Wang等[9]以DsRed为报告基因,建立一个有效的RNA干扰体系,在工业生产菌株康氏木霉中能维持稳定的干扰效果。尽管能有效地沉默靶基因,但由于表达shRNAs的质粒太大难以构建,难以同时沉默多个靶基因。本研究构建了质粒pPHL-Ppdc-shRNA-Tcbh1并转化到菌株DsRed-T.reesei中,此菌株是转入质粒pAN-Red1的重组菌株,由丙酮酸激酶基因的启动子pkil和cbh2终止子调控DsRed基因表达。另外,质粒pPHL-Ppdc-shRNATcbh1中的siRNA表达盒是由pPdc启动子和cbh1终止子调控表达。结果表明,DsRed的开放阅读框和siRNA表达盒都能通过PCR成功扩增,意味着DsRed基因和siRNA表达盒都完整地同源重组到菌株的基因组上。本研究构建了包含shRNA、Ppdc启动子和cbh1终止子的重组质粒。这个质粒比报道过的丝状真菌发夹RNA表达质粒更有效[25-27]。
在荧光显微镜上能清楚观察到菌丝发出红色荧光,相比对照组,红色荧光在筛选真菌干扰转化子时起到重要的标记作用(见图2)。结果表明,重组质粒pPHL-Ppdc-shRNA-Tcbh1所表达的shRNA能有效干扰靶基因的表达,报告基因DsRed为筛选干扰转化子起到至关重要的作用。本研究结果显示,干扰效率在DsRed-T3的转化子中高达86.96%,在DsRed-T12中为68.42%。干扰效果的不同可能有以下几个原因:第一,目标片段的位置不同。第二,转化子中可能发生非同源重组。Hoff等[28-30]发现在青霉菌中常常发生非同源重组,所以会产生不同的基因敲除菌株。第三,转化子中可能发生不同拷贝数的同源重组。Young等[31-32]发现质粒pAN7-1插入到单一位点上,却出现4~6个从头到尾串联排列的拷贝数。第四,丝状真菌中RNA依赖的RNA聚合酶(RdRP)对序列特异性的siRNAs的合成和扩增十分重要,为生物提供了源源不断的siRNA。此扩增数量的不同可能会导致基因沉默的效果不同[33-35]。利用粗糙脉孢菌qde-1基因产物为查询序列对T.reesei基因组进行blast,结果发现一个氨基酸同源性高达55%的RdRP。
不管是在真菌中还是在其他生物体系中,RAN干扰的高效率和序列特异性,报告基因DsRed的操作方便性和蛋白质组学与微阵列分析的不断改进,都极大促进了新型高通量手段的发展[36-38]。T.reesei中DsRed基因的高效沉默,加上高通量表达的筛选方式,有望加速我们对真菌生理过程的理解,有利于实现更多针对性的遗传操作以及对当前和未来工业利用真菌的持续优化。我们的数据清晰地表明,上述用于T.reesei的RNA沉默方法是有效的,DsRed基因相比于对照菌株通过发出红色荧光为筛选沉默转化子提供了更为方便的手段。这个体系在T.reesei中可能产生多重敲除的重组菌株,能研究涉及共同生物合成途径的多个基因的功能,并且能用于同时沉默多个靶基因。最近为了构建真菌RNAi的发夹载体文库,Gateway(Invitrogen)的体外重组技术用于高通量克隆[1]。本研究所改进的方法能用于构建包含siRNAs的载体继而沉默涉及代谢途径的多个靶基因,因此能同时沉默多个靶基因。为了能有效地同时沉默多个靶基因,短发夹RNA (shRNA)应筛选后插入到载体上。
本研究结果表明,重组载体表达的siRNA在菌株DsRed-T.reesei中能有效沉默DsRed基因,报告基因DsRed为筛选干扰重组菌提供了方便的手段。本文所述的研究方法可用于通过调控控制蛋白的表达研究同源或异源表达情况。
[1]Weld RJ,Plummer KM,Carpenter MA,et al.Approaches to functional genomics in filamentous fungi[J].Cell Res,2006,16(1):31-44.
[2]Meyer V.Genetic engineering of filamentous fungi-progress,obstacles and future trends[J].Biotechnol Adv,2008,26 (2):177-185.
[3]Martinez D,Berka RM,Henrissat B,et al.Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn.Hypocrea jecorina)[J].Nat Biotechnol,2008,26(5): 553-560.
[4]Fire A,Xu S,Montgomery MK,et al.Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans[J].Nature,1998,391(6669):806-811.
[5]Verdel A,Jia S,Gerber S,et al.RNAi-mediated targeting of heterochromatin by the RITS complex[J].Science,2004,303 (5658):672-676.
[6]Cogoni C1,Macino G.Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase[J].Nature,1999,399(6732):166-169.
[7]Simmer F,Buscaino A,Kos-Braun IC,et al.Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast[J].EMBO Rep,2010,11(2):112-118.
[8]Salame TM,Ziv C,Hadar Y,et al.RNAi as a potential tool for biotechnological applications in fungi[J].Applied Microbiology and Biotechnology,2011,89(3):501-512.
[9]Wang S,Xing M,Tian S,et al.Establishment of an efficient RNA silencing system in Trichoderma koningii using DsRed as a reporter[J].Folia Microbiol(Praha),2013,58(6):601-606.
[10]He R,Guo W,Wang L,et al.Construction of an efficient RNAi system in the cellulolytic fungus Trichoderma reesei[J].Microbiol Methods,2015,108:70-73.
[11]Barton LM,Prade RA.Inducible RNA Interference of brlA beta in Aspergillus nidulans[J].Eukaryot Cell,2008,7(11): 2004-2007.
[12]Kiiskinen LL,Kruus K,Bailey M,et al.Expression of Melanocarpus albomyces laccase in Trichoderma reesei and characterization of the purified enzyme[J].Microbiology,2004,150 (Pt 9):3065-3074.
[13]Wang B,Xia L.High efficient expression of cellobiase gene from Aspergillus niger in the cells of Trichoderma reesei[J].Bioresour Technol,2011,102(6):4568-4572.
[14]LarrondoLF,Colot HV,Baker CL,et al.Fungal functional genomics:tunable knockout-knock-in expression and tagging strategies[J].Eukaryot Cell,2009,8(5):800-804.
[15]Ruiz-Díez B.Strategies for the transformation of filamentous fungi[J].Appl MicrobiolBiotechnol,2002,92(2):189-195.
[16]Michielse CB,Hooykaas PJ,van den Hondel CA,et al.Agrobacterium-mediated transformation as a tool for functional genomics in fungi[J].Curr Genet,2005,48(1):1-17.
[17]Penttil¨a M,Nevalainen H,R¨att¨o M,et al.A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei[J].Gene,1987,61(2):155-164.
[18]Seiboth B,Hartl L,Pail M,et al.The galactokinase of Hypocrea jecorina is essential for cellulose induction by lactose but dispensable for growthon d-galactose[J].Mol Microbiol,2004,51(4):1015-1025.
[19]Nakayashiki H,Hanada S,Nguyen BQ,et al.RNA silencing as a tool for exploring gene function in ascomycete fungi[J].Fungal Genet Biol,2005,42(4):275-283.
[20]Hoff B,Kamerewerd J,Sigl C,et al.Homologous recombination in the antibiotic producer Penicillium chrysogenum:strain Delta Pcku70 shows up-regulation of genes from the HOG pathway[J].Appl Microbiol Biotechnol,2010,85(4):1081-1094.
[21]Armesto C,Maia FG,de Abreu MS,et al.Genetic transformation with the gfp gene of Colletotrichum gloeosporioides isolates from coffee with blister spot[J].Braz J Microbiol,2012,43 (3):1222-1229.
[22]Kalleda N,Naorem A,Manchikatla RV.Targeting fungal genes by diced siRNAs:a rapid tool to decipher gene function in Aspergillus nidulans[J].PLoS One,2013,8(10):e75443.
[23]Yang J,Nie L,Chen B,et al.Hygromycin-resistance vectors for gene expression in Pichia pastoris[J].Yeast,2014,31(4): 115-125.
[24]Mikkelsen L,Sarrocco S,Lübeck M,et al.Expression of the red fluorescent protein DsRed-Express in filamentous ascomycete fungi[J].FEMS Microbiol Lett,2003,223(1):135-139.
[25]Janus D,Hoff B,Hofmann E,et al.An efficient fungal RNA-silencing system using the DsRed reporter gene[J].Appl Environ Microbiol,2007,73(3):962-970.
[26]Kadotani N,Nakayashiki H,Tosa Y,et al.RNA silencing in the phytopathogenic fungus Magnaporthe oryzae[J].Mol Plant Microbe Interact,2003,16(9):769-776.
[27]Nakayashiki H.RNA silencing in fungi:mechanisms and applications[J].FEBS Lett,2005,579(26):5950-5957.
[28]Hoff B,Kamerewerd J,Sigl C,et al.Homologous recombination in the antibiotic producer Penicillium chrysogenum:strain Delta Pcku70 shows up-regulation of genes from the HOG pathway[J].Appl Microbiol Biotechnol,2010,85(4):1081-1094.
[29]Schmitt EK,Bunse A,Janus D,et al.Winged helix transcription factor CPCR1 is involved in regulation of beta-lactam biosynthesis in the fungus Acremonium chrysogenum[J].Eukaryot Cell,2004,3(1):121-134.
[30]Walz M,Kück U.Targeted integration into the Acremonium chrysogenum genome:disruption of the pcbC gene[J].Curr Genet,1993,24(5):421-427.
[31]Young C,Itoh Y,Johnson R,et al.Paxilline-negative mutants of Penicillium paxilli generated by heterologous and homologous plasmid integration[J].Curr Genet,1998,33(5):368-377.
[32]Nizam S,Singh K,Verma PK.Expression of the fluorescent proteins DsRed and EGFP to visualize early events of colonization of the chickpeablight fungus Ascochyta rabiei[J].Curr Genet,2010,56(4):391-399.
[33]Nicolas FE,Moxon S,de Haro JP,et al.Endogenous short RNAs generated by Dicer 2 and RNA-dependent RNA polymerase 1 regulate mRNAs in thebasal fungus Mucor circinelloides[J].Nucleic Acids Res,2010,38(16):5535-5541.
[34]Sijen T,Fleenor J,Simmer F,et al.On the role of RNA amplification in dsRNA-triggered gene silencing[J].Cell,2001,107(4):465-476.
[35]Makeyev EV,Bamford DH.Cellular RNA-dependent RNA polymerase involved in posttranscriptional gene silencing has two distinct activitymodes[J].Mol Cell,2002,10(6):1417-1427.
[36]Neumann B,Held M,Liebel U,et al.High-throughput RNAi screening by time-lapse imaging of live human cells[J].Nat Methods,2006,3(5):385-390.
[37]Wheeler DB,Carpenter AE,Sabatini DM.Cell microarrays and RNA interference chip away at gene function[J].Nat Genet,2005,37 Suppl:S25-30.
[38]Walter T,Held M,Neumann B,et al.Automatic identification and clustering of chromosome phenotypes in a genome wide RNAi screen by time-lapse imaging[J].J Struct Biol,2010,170(1):1-9.
ShRNAs Silencing DsRed Gene in the Filamentous Fungus Trichoderma reesei
LI Jie-xuan1,LIU Wen-li1,LIANG Zhi-cheng2,WANG Shao-wen2,LIANG Xiu-yi1,SHE Wei-yi1,LIU Gang1,TIAN Sheng-li1
(1.Coll.of Life Sci.&Oceanol.,Shenzhen Municip.Key Lab.of Micro-Genetic Engin.,2.Shenzhen Municip.Key Lab.of Oceano-Ecol.&Bio-Res.,Shenzhen Uni.,Shenzhen 518060)
The establishment of a red fluorescent protein(DsRed)in Trichoderma reesei as a reporter gene of RNA interference method is reported in this paper.Firstly,a plasmid pANRed1 that expressed DsRed was constructed to transform T.reesei QM9414,and obtained a strain of DsRed-T.reesei that resist against hygromycin and could steadily express DsRed.Secondly,pyruvate decarboxylase promoter and the cellobiohydrolase I terminator cbh I as original parts was cloned into vector pPHL,and constructed pPHL-Ppdc-Tcbh1 plasmid.According to the sequence of DsRed gene,a specific siRNA interference sequence and another siRNA with nonhomogenous sequence siRNA as negative control,were cloned into vector pPHL-Ppdc-Tcbh1 and obtained recombinant plasmid.Transform it into DsRed-T.reesei,and screened transformant on PDA plate containing 100 μg/mL of hygromycin and 250 μg/mL of phleomycin.The results showed that approximately 79%of transformant appeared red fluorescence silence phenomenon,among them some transformant DsRed’s expressed almost total inhibition.Fluorescence quantitative PCR and Western blotting analysis showed that DsRed gene expression was down regulated to different degrees.The above results suggested that the study on gene expression in T.reesei could be regulated with this method.
Q78
A
1005-7021(2016)04-0007-09
10.3969/j.issn.1005-7021.2016.04.002
国家自然科学基金项目(31070044);深圳市科技基础研究发展计划项目(ZYC201105130092A)
李洁璇女,硕士研究生。研究方向为基因表达与调控。E-mail:195378637@qq.com
刘雯莉女,硕士研究生。研究方向为基因表达与调控。E-mail:235410806@qq.com。李洁璇与刘雯莉为并列第一作者。
*通讯作者。男,教授,博士,硕士生导师。研究方向为基因表达与调控。E-mail:sltian@szu.edu.cn
2015-10-27;
2016-01-12
