水生生物DNA序列相似度的算法
2016-02-07于喆
于喆
(辽宁省海洋水产科学研究院,辽宁 大连 116023)
水生生物DNA序列相似度的算法
于喆
(辽宁省海洋水产科学研究院,辽宁 大连 116023)
本文提出DNA序列相似度指标,建立DNA序列比对算法。首先,将水生生物DNA样本序列与现有的基因库中的DNA序列逐项比对;其次,在满足特定阈值条件下,确认样本序列的分类。利用Java编程语言来实现算法,通过MonteCarlo模拟和实际应用,验证算法的有效性。理论结果表明:在满足特定阈值条件下,通过计算DNA序列相似度,能够确定数据库中与未知DNA样本序列最相似的序列,判断样本序列的分类。模拟实验、对比研究和应用结果表明:相似度指标算法在有效判别DNA序列的分类,提高DNA序列匹配成功率,降低程序复杂度等方面具有优良特征。
DNA序列相似度指标;水生生物;MonteCarlo模拟
基因组携带了构成生物体生命形式的全部信息,主要以DNA序列形式存在。DNA序列本质上是一种线性多聚脱氧核糖核苷酸,由碱基、戊糖及磷酸组成,碱基又进一步分为:腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T)。DNA序列所携带的生命体遗传信息由碱基顺序体现,不同生物体的DNA具有独特的碱基顺序,即所有DNA主链结构相同,只是4种碱基A、G、C和T的排列顺序不同。因此,观察主链上碱基的排列顺序就可以比较DNA序列;计算不同DNA序列之间的相似度,可以量化分析不同物种的DNA序列相似程度,推断出物种之间的亲缘关系。
随着人类基因组计划的发展,产生了海量的DNA数据信息,超出了现有计算机的处理水平。因此,设计合理的相似度指标和检验算法,识别和分析DNA序列,比较物种DNA序列同源性,对完善DNA序列数据库匹配功能,实现大规模数据分析以及克服现有计算机内存容量和计算能力的不足具有重要意义,国内外已对此进行了类似的研究[1,2]。
随着分子生物学技术的发展,水生生物物种判别的研究和应用逐渐从形态学深入到蛋白质和DNA水平[3,4]。目前,基因序列分析是判别物种最直接的方式[3],已经实现了对一些海洋物种的成功判定[5,6]。在基因序列研究中应用较多的是通过动态规划方法确定序列的相关程度,关于DNA序列相关性预测方法包括比较建模法[7-9],主要指同源结构预测,即面向有同源结构的DNA比较所应用的技术。同源结构预测模型可以判定序列同源性大于30%的序列[8]。另一类方法是基于统计序列特征进行定义,如计算两DNA序列间的欧式距离来判定DNA序列相似性[10],这类研究对计算复杂性要求较高,早期研究相对较少,随着信息技术和计算机快速发展,统计思想越来越多地应用于相似度研究,如基于LSH距离的时间子序列查询算法[11],这类算法结合了DNA序列距离的度量性质和序列自身特征,能够有效提高算法性能[12,13]。因此本文中,基于统计序列思想构建DNA序列相似度算法,用于研究水生生物种质基因库DNA序列的相似性。
首先,对数据库中的目标序列进行预处理,生成空间向量,并将向量数据储存在数据库中;然后,对待查DNA序列生成待查向量,通过计算相似度找出待查序列和目标序列间所有匹配程度超过一定阈值的序列片段对,确定数据库中与待查DNA样本序列最相似的序列,在满足特定阈值条件下,判断与样本序列最相似的DNA序列,确定样本序列的分类;最后,通过Java语言实现算法,应用于水产种质基因库信息平台,并使用MonteCarlo模拟和算法实际应用验证指标算法的有效性。
1 序列相似度算法
DNA序列特征包括内容和形式两方面[14],内容指碱基的含量,形式指其排列方式。通过综合考虑两种特征表达序列成分,分析序列碱基的含量和结构差异,主要是考察碱基关联方式的出现频率进行比对:首先,确定连接方式的步长,构建向量空间模型;然后,按步长对两条DNA链进行整理,以碱基关联方式为基底生成两个多维向量;最后,计算两个向量余弦相似度指标,即相似度值,确定待查序列和目标序列间所有匹配程度超过一定阈值的序列片段对,找出数据库中与未知DNA样本序列最相似的序列,在满足特定阈值条件下,能够确定与样本序列最相似的DNA序列,判断样本序列的分类。
第一步:构建向量空间模型。在该模型中,每个对象映射为一个特征向量,首先,通过确定连接方式的步长确定空间向量的基底。记待查询序列长度为L0,数据库样本数为N,样本长度为Lt,t=1,A,N,代表序列编号,其中每一种编号对应一种水生生物。
确定步长满足d=d0,如在d0=2时,即统计A、T、G、C两两组合在序列中的数量。表1为A、T、G、C两两组合统计表,由A、T、G、C两两组合生成空间基底总数即为n=4d0。在d0=2时,对应基底总数即为n=16,即为表1中的总列数。

表1 A、T、G、C两两组合Tab.1 Pairwise Combination of‘ATGC’
第二步:按步长对DNA链进行整理,以碱基关联方式为基底生成多维向量,按照每种基底的频数生成空间向量,则查询序列即:
v(L0)=(W1(L0),A,Wn(L0))1×n
数据库样本Lt对应的空间向量为:
v(Lt)=(W1(Lt),A,Wn(Lt))1×n
第三步:计算两个向量余弦相似度指标,即相似度的值。

第四步:将序列按相似度大小排序,判断数据库中与样本DNA序列最相似的序列^L。
第五步:计算序列^L与数据库中其他N-1个序列的余弦相似度指标,确定其中最大值记为序列^L对应的阈值M。
第六步:判断样本序列L0与^L相似度与序列^L对应的阈值M大小,满足相似度大于阈值,才能确认样本序列L0与^L相似度具有统计意义。
从以上指标设计的步骤中可以看出,这种方法所得到的相似度能够在一定的步长下计算出DNA相似程度,整体波动范围为0~1。同时,可以进一步修改设定不同的步长值满足d=d0(如d0=3、4、A),对序列相似度进行补充说明。但是,由于方法没有考虑DNA实际空间结构,更多是基于频率,会造成整体比对相似度偏高。
2 模拟实验与程序实现
为了满足DNA序列查询功能的基本要求,笔者采用MonteCarlo模拟实验,基于水生生物种质基因库资源平台系统方案设计,通过对水生生物种质基因库资源碱基配对统计,按照相应碱基或者碱基组合比例随机生成样本,选取数据库中已有DNA序列进行加工。
随机抽取数据库中的一条序列,用本文所提出的相似度指标比对算法进行查询,计算最佳匹配序列的相似度指标,并在序列基础上随机改变5%、20%、50%、100%(完全随机生成新DNA序列),依次进行对比。共进行500次随机抽取操作,取所计算的平均值为最终结果。为了进一步说明本文算法的可靠性和实用性,区分步长d=2、d=3、d=4时,通过构建不同基底进一步计算相似度指标,表2为不同步长的相似度实验结果。
从表2可以得出以下两个结论:
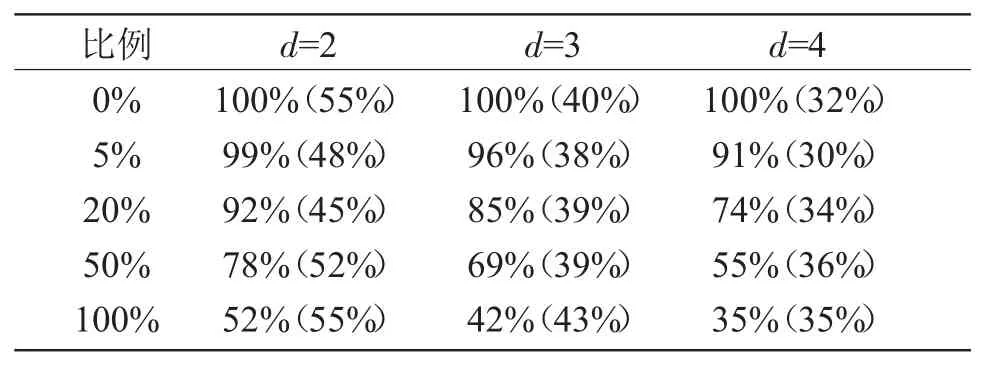
表2 不同步长的相似度实验结果Tab.2 Similarity Test Results under Different Steps
(1)随着步长增加,序列相似度下降,对应阈值也有所下降。
(2)随着待查询样本序列中同源性降低(随机部分增加),序列相似度下降,阈值变化不明显。
实验结果说明:判定序列相似度必须结合步长和相似度,阈值限定对非同源性样本序列筛除具有显著效果。参考MonteCarlo模拟实验结果,考虑在5%的容错机制下,步长d=2、3、4时,必须保证相似度在99%、96%和91%以上。
为了比较本文提出的序列对比算法的意义,与另外一类序列比对研究中常用的BLAST算法进行对比,结合数据模拟实验进行验证。
(一)算法分析和数据模拟实验表明两类算法的时间复杂度。Blast的核心算法是对两个满足长度相等,且形成无空位完全匹配的DNA序列的子序列,首先找出待查序列和目标序列间所有匹配程度超过一定阈值的序列片段对,然后根据给定的相似性延伸阈值,得到一定长度的相似性片段。Blast算法本质上是一类动态规划算法,通过定义变量(包括得分矩阵和罚分矩阵),计算最优局部比对,确定最佳对位排列,帮助人们做出最佳选择,但是由于源序列中大量子序列需要和待比对序列索引表所有子序列进行比较,计算步骤多,速度较慢。假设数据库中的目标序列和待查序列的长度为L1与L2,则序列比较的时间复杂度为O(L1L2)。在本文提出的序列比对算法中,首先,对数据库中的目标序列进行预处理,确定由不同长度的基底生成相应的特征向量。每个目标序列,如L1确定特征向量的时间复杂度为O(L1),不占用实际DNA序列对比的时间,特征向量数据储存在数据库中;然后,对待查序列确定其待查特征向量,通过计算相似度找出待查序列和目标序列间所有匹配程度超过一定阈值的序列片段对,时间复杂度为O(L2),有效降低了在实际比对中耗费的时间。在数据实验中,随机抽取数据库中的一条序列,分别使用本文所提出的相似度比对算法和Blast算法进行查询,进行DNA序列比对,查找序列分类,计算耗费时间。实验结果显示:Blast算法耗时42.81s,本文相似度比对算法耗时24.17s,有效控制了算法的时间复杂度。
(二)通过数据实验进行小片段序列在基因组中搜寻定位。首先,随机抽取数据库中的一条序列,分别采用完整方式和间隔方式抽取目标序列80%、60%和40%的片段;然后,用本文所提出的相似度比对算法和Blast算法进行查询,进行DNA序列比对查找序列分类。结果显示:若序列足够完整,即序列包含充分的生物基因的信息,无论是采用完整方式或是间隔方式抽取局部序列,本文所提出的序列比对算法更为有效。对按照完整方式抽取目标序列小片段(25%),Blast算法的准确性有一定优势,本文相似度比对算法计算最佳匹配的相似度指标和相应的阈值,同样可以说明目标序列相似度的可信度,具有一定的参考价值。对不同物种同源序列的搜索,为了说明算法的有效性,寻找可信度较高的目标序列,同样需要保证序列包含充分的生物基因信息。
(三)通过数据实验对基因组大片段序列进行共线性分析。在具体数据实验中,随机抽取数据库中的一条序列,采用完整和间隔两种方式随机改变5%、10%、20%的片段,计算最佳匹配的相似度指标和相应的阈值,结果可以得到类似的结论:若序列能够包含充分的生物基因信息,无论是采用完整方式或是间隔方式抽取局部序列,则本文所提出的序列比对算法都更为有效,说明本文算法具有一定的参考价值。
本文用JAVA语言实现了此算法并作为功能模块运行在辽宁省水产种质基因库信息平台上,图1、图2为程序运行截图。图1中录入物种的基因序列片段并选取相应的阈值、片段类型、物种种类等参数。图2为此基因序列片段在数据库中的比对后,按照相似度进行了打分并排序。平台能够对不同物种的DNA序列进行比较,说明相似度算法能够计算出目标序列相似度,具有较强参考价值。

图1 向系统输入序列片段Fig.1 Input Sequence Fragment to the System

图2 输出序列查询的结果Fig.2 OutputThe Results of the Query Sequence
3 讨论
研究发现,本文提出的相似度算法可能损失了DNA空间部分连接信息,导致结果有误差。若研究对象只是DNA序列的小片段,由于信息量不足而导致相似度与实际情况差别较大,无法得到目标序列,需要结合其他算法予以说明。但总体上,若序列足够完整,即序列包含充分的生物基因信息,无论采用完整方式或是间隔方式抽取局部序列,本文所提出的序列比对算法都有效。本方法所得到的结果比较符合生物物种的进化规律,对研究物种的同源性有一定价值。
水生生物的分类目前仍然主要依赖于形态学特征,然而随着样本序列的复杂性提高,传统的比对算法对序列片段的限制条件和时间复杂度较高,已逐渐无法满足物种鉴定需求。本文从统计学特征出发,通过设计DNA序列比较算法确定物种分类,在序列包含充分的生物基因信息时,序列比对算法都有效,具有较强的实用价值,在保护海洋生物学及生物多样性调查等领域中有较好的应用前景。目前国内水生生物DNA序列比对算法研究还不多,数据库平台的DNA序列样本不足,制约了相关研究的进展。随着全球气候变化、生态环境等问题的日益严峻,人类对理解生物多样性的要求日益迫切,物种的准确和快速鉴定对生物多样性资源的保护有重要意义。
[1]LiW.ArespectralanalysisusefulforDNAsequence analysis[J].DNA in Chromatin,At the Frontiers of Biology,Biophysics,andGenomics,Arcachon,France,2002(3):23-29.
[2]张宝华,王海水,许禄.DNA序列编码及相似度计算[J].高等学校化学学报,2006(12):2277-2280.
[3]孙超,苏彦平,刘洪波,等.水生生物近缘种和产地的分子生物学判别[J].水产学杂志,2011,24(3):53-59.
[4]姜维,王启军,邓捷,等.以川陕哲罗鲑为目标物种的水样环境DNA分析流程的优化[J].应用生态学报,2016,27(7):2363-2371.DOI:10.13287/j.1001-9332.201607.015.
[5]Steinke D,Zemlak T S and Hebert P D N.Barcoding nemo: DNA-based identifications for the ornamental fish trade[J]. Plosone,2009,4(7):1-5.
[6]Kartavtsev Y P,Park T J,Vinnikov K A,et al.Cytochromeb(cyt-b)gene sequence analysis in six flatfish species(Teleostei,Pleuronectidae),with phylogenetic and taxonomicinsights[J].MarBiol,2007,152(4):757-773.
[7]郝柏林,张淑誉.生物信息学手册[M].上海:上海科学技术出版社,2000.
[8 Sali A and Blundell T L.Comparative protein modeling by satisfaction ofspatial restraint[J].J MolBiol,1993,234(3): 779-815.
[9]赵东明,强小利,刘向荣.一种蛋白质结构同源建模的DNA算法[J].北京大学学报:自然科学版,2009,45(5):748-752.
[10]钱能,金文东.DNA序列比对分析中的统计特征方法[J].浙江工业大学学报,2005,33(2):173-175.
[11]汤春蕾,董家麒.基于LSH的时间子序列查询算法[J].计算机学报,2012,35(11):2228-2236.
[12]戴东波,汤春蕾,邱伯仁等.一种优化多重过滤的序列查询算法[J].计算机研究和发展,2010,47(10):1785-1796.
[13]廖丽,伍绍佳.优化多重过滤的序列查询算法研究[J].网络安全技术与应用,2014(6):104-105.
[14]JoaoSand JoaoM.Introduction tocomputational molecular biology[M].Brooks/Cole PublishingCompany:a Division of ThomsonLearning,1997.
Comparison Method of DNA Sequence Similarity in Aquatic Organisms
YU Zhe
(Liaoning Ocean and Fisheries Science Research Institute,Dalian 116023,China)
DNA sequence similarity index was proposed and the comparison method was constructed in this paper.Firstly,DNA sample sequence of aquatic organisms was compared with DNA sequence in the existing gene bank;secondly,sample sequence classification was determined based on the specific threshold condition.The algorithm was implemented using Java programming language,and the validity of the algorithm was verified by MonteCarlo simulation and application.Theoretical results showed that by calculating DNA sequence similarity index,the most similar DNA sequence in the gene bank of the unknown sample of DNA sequence was determined under certain threshold condition.Simulation experiments,comparison research and application results showed that Comparison Method of the similarity index was effective for classifying DNA sequence,improving matching rate of DNA sequences and reducing complexity of the program.
DNA sequence similarity index;aquatic organisms;MonteCarlo simulation
S917
A
1005-3832(2016)05-0022-05
2016-04-19
国家水产种质资源平台项目(2016DKA30470);辽宁省水产种质基因库信息平台建设(201519).
于喆(1984-),男,硕士,工程师,从事生物信息技术研究.E-mail:chinayuzhe@126.com
