噁唑霉素及其类似物的合成研究进展
2016-01-31梁大伟赖巴特
梁大伟,赖巴特,苏 静
(1. 雅安职业技术学院 药学检验系,四川 雅安 625000; 2. 安斯泰来制药(中国)有限公司,辽宁 沈阳 110041;
3. 东北制药集团沈阳第一制药有限公司,辽宁 沈阳110141)
噁唑霉素及其类似物的合成研究进展
梁大伟1*,赖巴特2,苏静3
(1. 雅安职业技术学院 药学检验系,四川 雅安 625000;2. 安斯泰来制药(中国)有限公司,辽宁 沈阳 110041;
3. 东北制药集团沈阳第一制药有限公司,辽宁 沈阳110141)
摘要:噁唑霉素及其类似物是由一些链霉菌产生的抗生素,它们都由3个基本骨架单元连接,其独特的生物活性和新颖的化学结构吸引了越来越多的化学与药物化学研究者的关注,从最初发现至今已经有很多研究小组报道了相关的合成研究成果. 本文作者基于3个基本组成结构单元,综述了其合成的研究进展,分析了每种合成方法所采用的关键策略,并对该类药物的全合成前景进行了展望.
关键词:噁唑霉素;抗生素;合成;研究进展
噁唑霉素A (1) 与Neooxazolomycin (2) 是1985年由日本名古屋大学、静冈大学与万有制药公司的科研人员联合从一些链霉菌中分离得到的抗生素[1-2],随后其他类似物如噁唑霉素B (3) 与噁唑霉素C (4)[3]、16-Methyloxazolomycin (5)[4-5]、Curromycin A (6) 与Curromycin B (7)[6]、KSM-2690 B (8) 与KSM-2690 C (9)[7]陆续在链霉菌中被发现,这些化合物的结构如图1,相应药理学研究显示这类化合物具有潜在而广谱的抗菌作用及体外抗肿瘤活性,能够有效地抑制人体细胞中牛痘、I型疱疹病毒与甲型流感病毒的增殖,此外,小鼠实验显示该类药物毒性较低 (LD50= 10.6 mg/kg). 在结构特征上,这类化合物通过三烯链与(E,E)-二烯链将噁唑端位侧链与特异螺旋β-内酯/γ-内酰胺连接起来,结构差异主要在于噁唑环上取代基的不同、三烯链上双键的顺反异构与内酯环的差异. 从发现至今的三十年里,关于这类化合物的全合成或结构片段(片段A、B与C) 的合成方法学研究报道较多,已经成为有机化学家和药物化学家特别关心的研究课题之一,新的全合成路线也不断出现. 为此,本文作者对噁唑霉素及其类似物的一些主要合成研究进行了综述.

图1 噁唑霉素及其类似物的结构Fig.1 The structures of oxazolomycin and its analogs
1噁唑霉素及其类似物的全合成
从发现至今,噁唑霉素及其类似物的全合成鲜见报道. 1990年,英国罗切斯特大学的KENDE等[8]首次完成了Neooxazolomycin (2) 的立体选择性全合成;随后日本长崎大学的HATAKEYAMA等[9]在2007年也完成了对Neooxazolomycin (2) 的全合成,此外,该小组于2011年还报道了对噁唑霉素A (1) 的全合成[10]. 有很多研究小组对噁唑霉素及其类似物的全合成进行了研究,其中对于这类化合物的结构片段A、B与C的合成研究较多.
1.1 片段A的合成
1989年,KENDE等[11]首次高效对映立体选择性地合成了片段A中C-1′—C-5′部分α,α-二甲基-β-羟基羧酸酯 (14) (见图2).

图2 KENDE等对噁唑霉素的片段A中α,α-二甲基-β-羟基羧酸酯的合成[11]Fig.2 KENDE′s synthesis of α,α-dimethyl-β-hydroxy acid ester of fragment A in oxazolomycin[11]
以Z式烯炔醛10与手性试剂11在路易酸SnCl2作用下经过Aldol加成、重排、非对映选择性得到R,S-12 (de> 99%),双键的顺式构型保持不变;然后在强碱作用下水解,羧基甲酯化得到酯13 (ee> 99%);最终,酯13在硅醚化试剂TBSOTf及碱,臭氧裂解作用下得到光学纯的α,α-二甲基-β-羟基羧酸酯 (14),其立体结构以手性Seebach二酯通过简单衍生化获得的同样产物14进行对照验证[12]. 该方法只构造了手性α,α-二甲基-β-羟基羧酸酯片段,过程较复杂,但仍为噁唑霉素的全合成奠定了重要基础.
MOLONEY课题组[13]在2002年报道采用区域选择性Stille偶联反应为关键步骤合成了噁唑霉素B中片段A的三烯结构片断(图3). 醇15经过Swern氧化得到醛16;酯17依次经过溴代、膦酸酯化得化合物18;醛16与膦酸酯18在碱NaH作用下发生Wadsworth-Emmons烯化反应得到单一化合物19,进一步碘代烯烃化合物经过Stille偶联反应得到目标(E,E,E)-三烯结构片段20b. 在此基础上,作者用同样的策略分别合成了噁唑霉素A与C中片段A的三烯侧链25b与26b(图4). 该课题组于2003年[14]采用同样的策略合成了噁唑霉素A、B与C中的片段A,详细叙述了该反应过程,并考察了三烯结构片段中Z与E式的异构化. 该方法对噁唑霉素中片段A的三烯侧链进行研究,合成路线操作简单,对于噁唑霉素的合成具有潜在应用价值,可有效地应用于噁唑霉素活性衍生物的合成中.
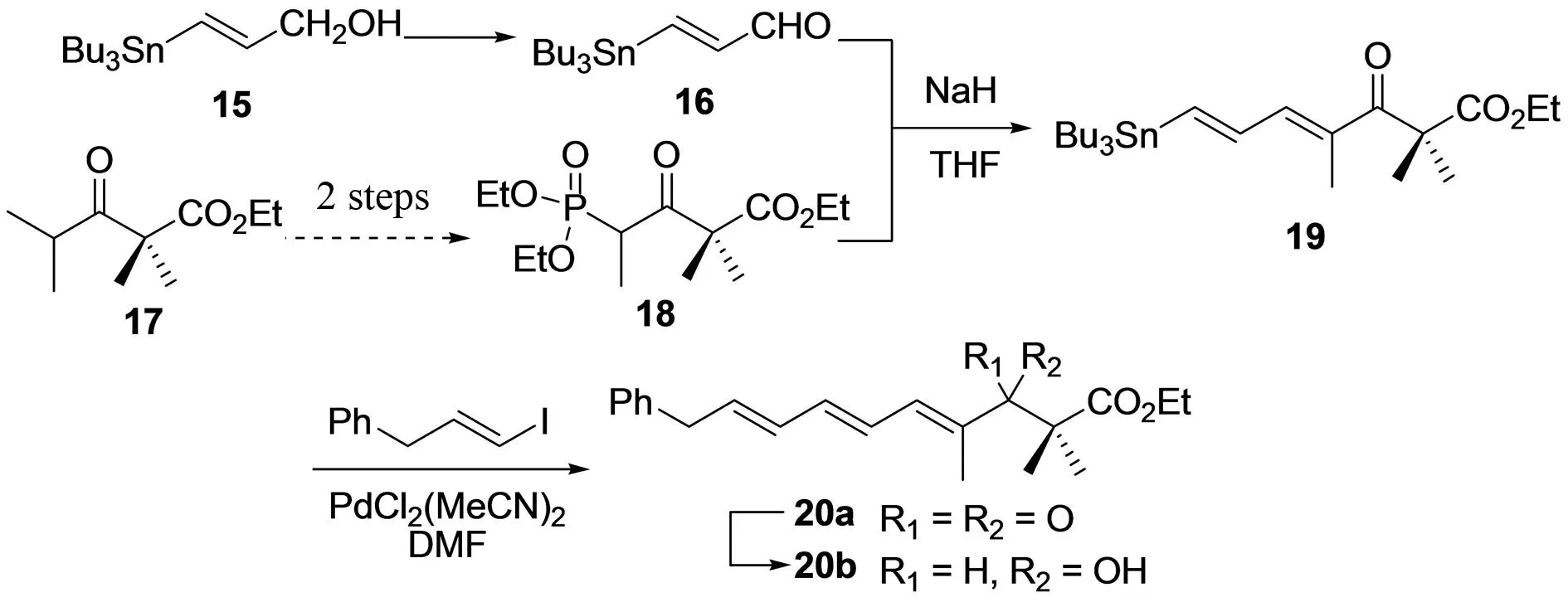
图3 MOLONEY等对噁唑霉素B的片段A中三烯的合成[13]Fig.3 MOLONEY′s synthesis of the triene of fragment A in oxazolomycin B[13]

图4 MOLONEY等对噁唑霉素A与C的片段A中三烯的合成[13]Fig.4 MOLONEY′s synthesis of the triene of fragment A in oxazolomycin A and C[13]
1.2 片段B的合成
MOLONEY课题组在完成噁唑霉素的片段A的合成研究后,又相继报道了片段B的立体选择性合成研究[15](图5). 以丙烯醛27为原料,与1-溴-2-丁烯在三价铬与四氢铝锂的催化作用下发生Hiyama反应得到烯丙醇衍生物28,其立体化学通过二维核磁与分子模拟确定;进一步对烯丙位的羟基进行硅醚保护得化合物29,然后采用Andrus合成法[16],在催化剂AD-mix-α作用下对化合物29端位烯键进行双羟化得到比例为1∶1的非对映异构体30,高碘酸钠可将邻位二醇氧化裂解成醛,进一步使用硼氢化钠还原得到醇31,最后采用Stille偶联反应成功合成噁唑霉素的片段B (32). 该方法采用成熟的Hiyama反应与Stille偶联反应为关键步骤,且其他反应步骤也较简单,具有较好的实际应用价值.

图5 MOLONEY等对噁唑霉素的片段B的合成[15]Fig.5 MOLONEY′s synthesis of fragment B in oxazolomycin[15]
1.3 片段C的合成
1996年,MOLONEY等[17]以简单的L-丝氨酸甲酯 (33) 为原料,采用Seebach合成法[18],与特戊醛在碱三乙胺作用下生成噁唑烷34,然后在缩合剂EDAC、DMAP作用下与相应的羧酸反应生成主要的,可分离的顺式非对映异构体酰胺35与36,进一步在碱性条件下发生分子内Aldol反应得到特异螺旋β-内酯/γ-内酰胺37-40,其中化合物37与39的立体构型与噁唑霉素的结构片段C的立体构型相同 (图 6). 此方法以廉价易得的原料,经过简单的化学反应成功合成噁唑霉素的结构片段C,但存在噁唑烷与特异螺旋β-内酯/γ-内酰胺的非对映异构体的分离问题. 在此基础上,该课题组于2014年报道了以L-苏氨酸为原料,采用非对映选择性分子内Aldol反应策略,成功构建了系列螺旋β-内酯/γ-内酰胺衍生物,虽然产率较低,但成功获得了光学纯度较高的β-内酯/γ-内酰胺类非对映异构体,对于噁唑霉素的结构片段C的改造具有重要意义[19].
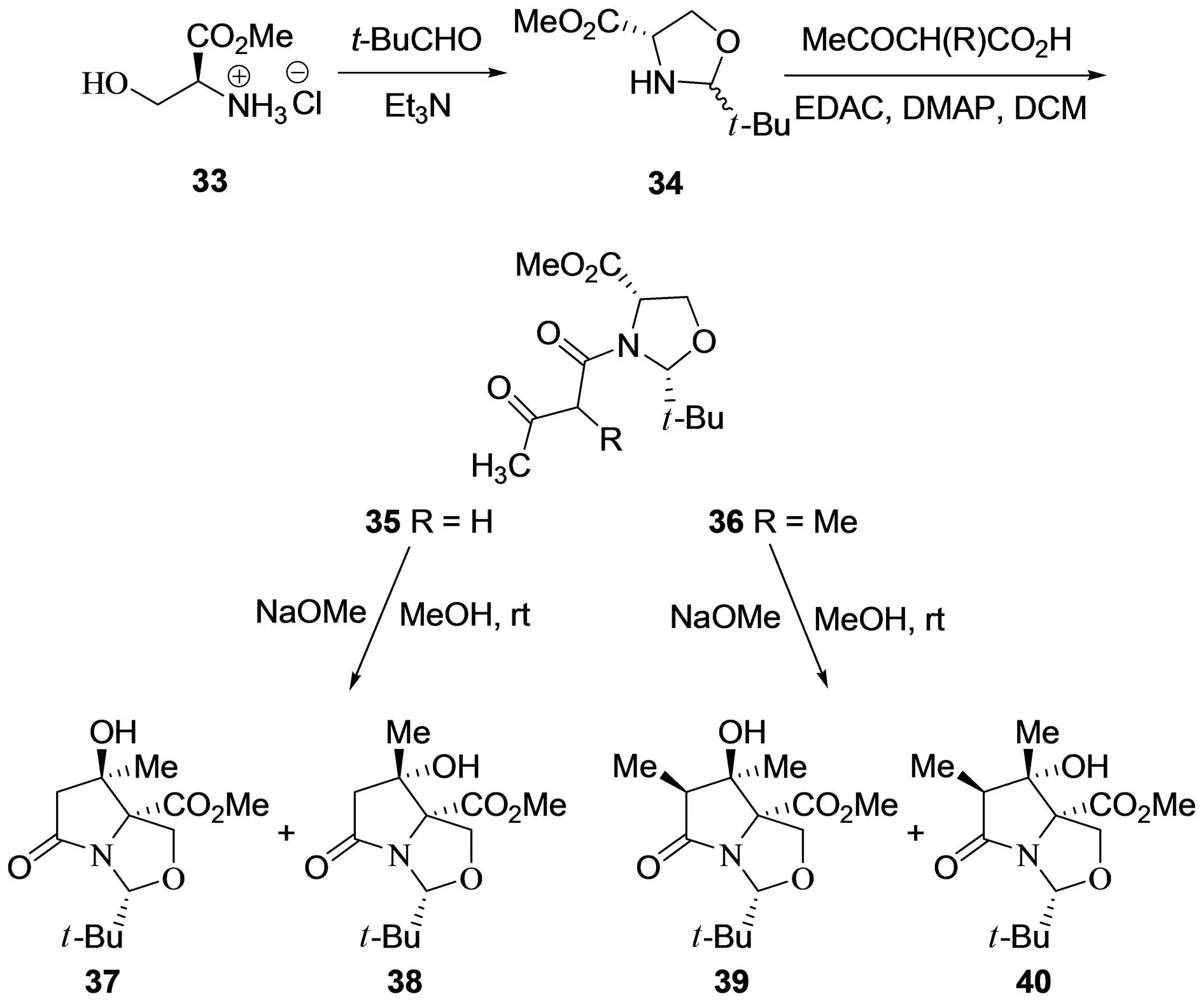
图6 MOLONEY等对neooxazolomycin的片段C的合成[17]Fig.6 MOLONEY′s synthesis of fragment C in neooxazolomycin[17]
TAYLOR课题组于2000年采用手性的L-脯氨酸 (41) 为起始原料[20],首先与新戊醛在酸性条件下反应生成单一非对映异构体的五元环状中间体42,同时保护羧基与氮原子;然后在有机碱LDA作用下,羰基α位引入醚侧链得化合物43,进一步在酸性条件下脱去第一步保护基新戊醛得到化合物44,同时采用Boc将氮原子保护得45,催化加氢脱去侧链的苄基得化合物46,在改进的Mitsunobu反应条件下,发生分子内成酯反应得酯47,最后采用Sharpless合成法[21],将氮原子的α位亚甲基氧化成羰基得到对映选择性的β-内酯/γ-内酰胺48 (图 7). 串联Aldol反应与内酯化反应为此条路线的关键反应步骤,原料简单易得,反应步骤易于操作,为噁唑霉素的片段C的合成提供了一种重要方法,其中只有最后一步氧化的产率较低,有待进一步改进.
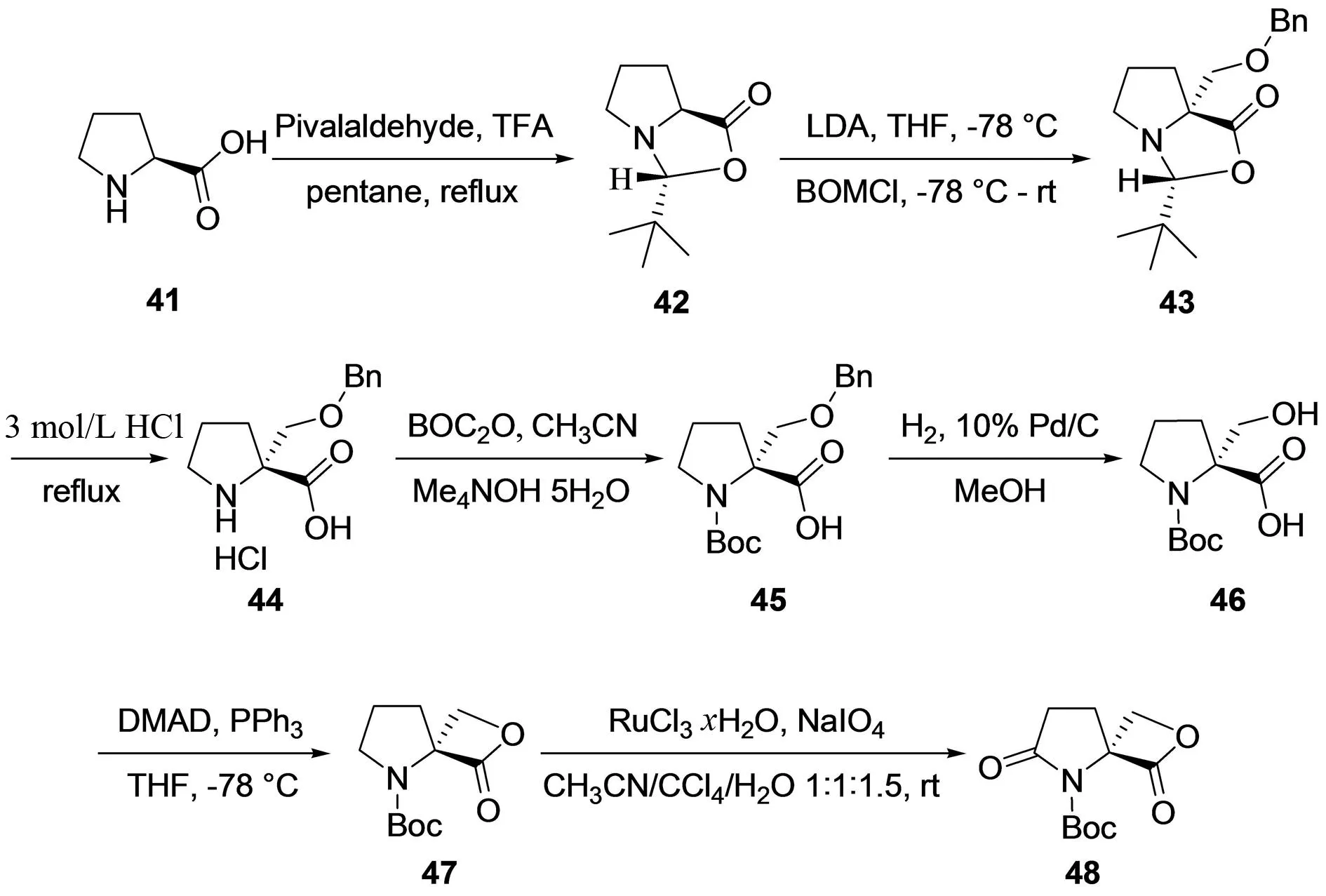
图7 TAYLOR等对噁唑霉素的片段C的合成[21]Fig.7 TAYLOR′s synthesis of fragment C in oxazolomycin[21]
噁唑霉素及其类似物的合成研究主要集中于其结构片段C的构造,2006年,MOHAPATRA等[22]报道了一种新颖的β-内酯/γ-内酰胺的合成方法(图8),以手性的噁唑烷酮49与(R)-(-)-3-Boc-2,2-二甲基噁唑啉-4-甲醛50为原料,在Crimmins改进法条件下[23],经过Evans-Aldol反应[24]得到高非对映选择性产物51,进一步用硼氢化钠还原脱去噁唑烷形成二醇52,随后的保护与脱保护策略选择性的使用苄基保护二级醇羟基得54,再将侧链一级醇羟基甲磺酸酯化得化合物55,然后在对甲苯磺酸-甲醇体系中,噁唑啉开环,氨基Boc保护部位与磺酸酯成环形成新的五元吡咯烷中间体56. 在成功构造五元吡咯烷后,下一步重点为氮原子两侧引入羰基与四元内酯. 首先化合物56经过IBX-DMSO-THF氧化,然后与甲醛反应得二羟甲基衍生物57;为了保护二羟甲基结构片段,对其进行保护得异亚丙基衍生物58,随后经过四氧化钌选择性氧化得吡咯烷酮中间体59,酸性脱保护得二醇60,二醇选择性硅醚保护得化合物61,然后在高碘酸钠与三氯化钌作用下,将未保护的羟甲基氧化为羧基得62,进一步弱酸性条件脱硅醚保护而裸露醇羟基,最后在Mitsunobu条件下[25],分子内成酯,成功构建了四元环内酯的螺环中间体64,即噁唑霉素及其类似物的结构片段C. 此合成方法为噁唑霉素及其类似物的结构片段C的合成提供了一种全新的策略,但反应步骤较长,且多步反应条件较苛刻,实际应用具有一定局限性.

图8 MOHAPATRA等对噁唑霉素的片段C的合成[22]Fig.8 MOHAPATRA′s synthesis of fragment C in oxazolomycin[22]
以链状小分子为起始原料合成噁唑霉素的片段C还见于PATTENDEN等[26]的报道中(图9),作者以烯炔醇65为原料,首先对其双键进行Sharpless环氧化,然后在(-)-酒石酸二异丙酯、钛酸四异丙酯与过氧化氢异丙苯作用下构造手性中心,并进一步在DBU作用下,与三氯乙腈反应生成相应的亚胺环氧化合物66;化合物66在AlEt2Cl作用下发生环合反应生成噁唑啉67,对其羟基进行硅醚保护得化合物68,进一步稀盐酸水解得α-乙炔基胺69,并立即与2-溴丙酰基氯反应得到化合物70. 为了便于构造内酯四元环,下一步需将一级醇转变为酸基或酯基结构片断,作者采用TPAP与NMO先将醇羟基氧化为醛基,进一步经过NaClO2与NaH2PO4氧化为羧基得71,重氮甲烷对酸71进行甲酯化得酯72;甲酯72在Bu3SnH与AIBN作用下,羰基α-溴代位置与炔基成环生成主要的非对映异构体73,最后OsO4-TMEDA对环外双键进行双羟化得吡咯烷酮74,可作为合成噁唑霉素A (1) 与Neooxazolomycin (2)的片段C的前体. 关于γ-内酰胺的构造方法较多,如Michael加成-还原内酰胺法[27],串联反应法[28]等. 该方法以非手性小分子链状物为起始原料,对于立体选择性合成噁唑霉素的结构片段C具有重要方法学意义.

图9 PATTENDEN等对噁唑霉素的片段C的合成[26]Fig.9 PATTENDEN′s synthesis of fragment C in oxazolomycin[26]
2008年,OHFUNE等[29]以手性的3,4-二取代焦谷氨酸衍生物75为原料,经过两步简单反应得到N-甲基取代的叔丁酯76,然后催化加氢脱去苄基保护基,并进一步与氯甲酸甲酯反应得中间体77,强有机碱作用下,在化合物77的叔丁酯羰基α-位引入羧基得到化合物78a,同时伴随形成五元内酯产物78b;化合物78b经过还原开环,醇羟基部位硒苯醚化,羧基重氮甲酯化得化合物79,硒苯醚基团在双氧水氧化条件下转变为双键产物80,最后,采用Zn(BH4)2选择性还原叔丁酯基团得化合物81,可作为合成噁唑霉素A (1) 与Neooxazolomycin (2)的片段C的前体(图10). 该方法提出了一种全新的合成噁唑霉素的结构片段C的策略,但原料来源有限,对于噁唑霉素的全合成具有一定的局限性.
MONDAL等[30]在之前MOHAPATRA合成法的基础上,以交叉Cannizzaro反应与双立体差异Crimmins-Aldol反应为关键步骤合成螺环β-内酯/γ-内酰胺结构(图11).
首先,手性辅助试剂82与具有相似手性五元环结构的3-Boc-2,2-二甲基噁唑啉-4-甲醛83在四氯化钛催化下发生Crimmins-Aldol反应生成84,然后经过六步常规反应构建五元吡咯烷中间体85;化合物85中的一级醇羟基在IBX-DMSO-THF反应条件下氧化为醛86,然后在碱性条件下,与甲醛在C-2位发生交叉Cannizzaro反应生成高非对映选择性的氨基甲酸内酯88,同时伴随少量二羟甲基衍生物87,化合物87在碱性条件下易于转变为氨基甲酸内酯88,后者经过Swern氧化将未成酯的一级醇羟基氧化为醛89,进一步经过常规反应转变为相应的羧酸甲酯90,然后在碱性条件下,双酯水解后,成功构建螺环β-内酯所需的结构片断中间体91,最后脱去Boc保护基及内酯化得化合物93,成功合成了噁唑霉素的片段C. 该方法与Mohapatra合成法的主要不同在于β-内酯的构建策略,以交叉Cannizzaro反应巧妙引入并保护合成β-内酯结构片段所需的羟甲基,具有重要方法学意义,但同样存在反应步骤较长,且多步反应条件较苛刻,实际应用具有一定局限性.
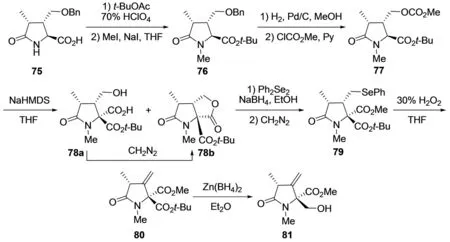
图10 OHFUNE等对噁唑霉素的片段C的合成[29]Fig.10 OHFUNE′s synthesis of fragment C in oxazolomycin[29]
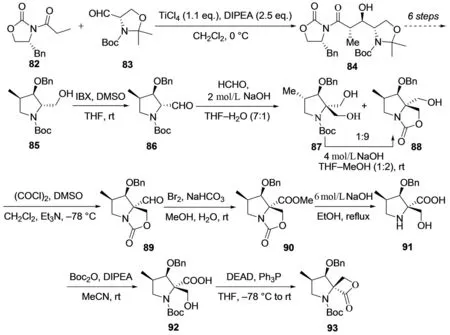
图11 MONDAL等对噁唑霉素的片段C的合成[30]Fig.11 MONDAL′s synthesis of fragment C in oxazolomycin[30]
2012年,DONOHOE等[31]以前手性的吡咯衍生物94为原料,不对称合成了螺环β-内酯/γ-内酰胺结构片段,同时包含连接结构片段C的侧链(图12). 首先,吡咯衍生物94经过部分Birch还原,与特戊酰碘甲酯连接,进一步还原二酯基团得二醇95,其具有γ-内酰胺的基本碳骨架;为了选择性的还原五元吡咯烷上的烯基双键,首先对二醇进行保护得吡咯啉衍生物96,进一步经硼烷还原,氧化得较高的区域选择性与非对映选择性的羟基吡咯啉衍生物97,邻位甲基的空间位阻可以很好地控制羟基的空间构型;戴氏-马丁试剂将羟基氧化为羰基,然后与烯丙基格氏试剂反应引入烯丙基侧链衍生物98,由于邻位甲基位阻效应,化合物98的立体选择性同样较高,进一步对烯位双键进行Grubbs II催化剂氧化与臭氧氧化裂解得α-羟基醛99;然后利用醛基易于衍生化的特点引入手性侧链得到理想的单一非对映异构体二醇衍生物100,再在Meerwein试剂作用下对侧链上的羟基甲醚化,臭氧氧化裂解侧链双键,还原乙酰化成酯101,氮原子邻位亚甲基经RuO2氧化羰基化,进一步氮脱Boc保护并甲基化得化合物102;化合物102在高浓度三氟醋酸作用下脱去MOM保护基,羟甲基经连续两步氧化过程转变为羧基衍生物103,对化合物103中羧基进行硅烷酯保护,侧链乙酸酯水解,Swern氧化得醛104;最后采用Nozaki-Hiyama-Kishi偶联反应策略,化合物104与碘代烯烃衍生物偶联,进一步被戴氏-马丁试剂氧化,(S)-2-甲基-CBS-氧杂硼啶作用下BH3·SMe2非对映选择性还原得到较高非对映选择性产物105,侧链硅酯保护基易于脱保护及分子内成酯构建β-内酯结构片段,可作为噁唑霉素A的合成原料. 该方法有效的不对称合成了噁唑霉素A中含有复杂侧链的吡咯烷酮结构片段,但仍然存在反应步骤较长,使用试剂较特殊等缺点,对实际噁唑霉素的全合成具有一定的限制性.
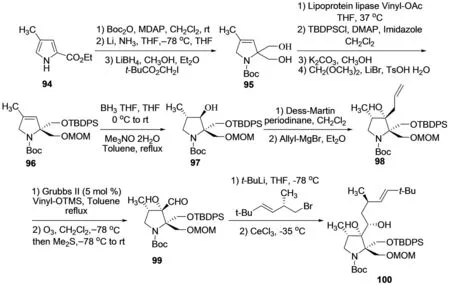
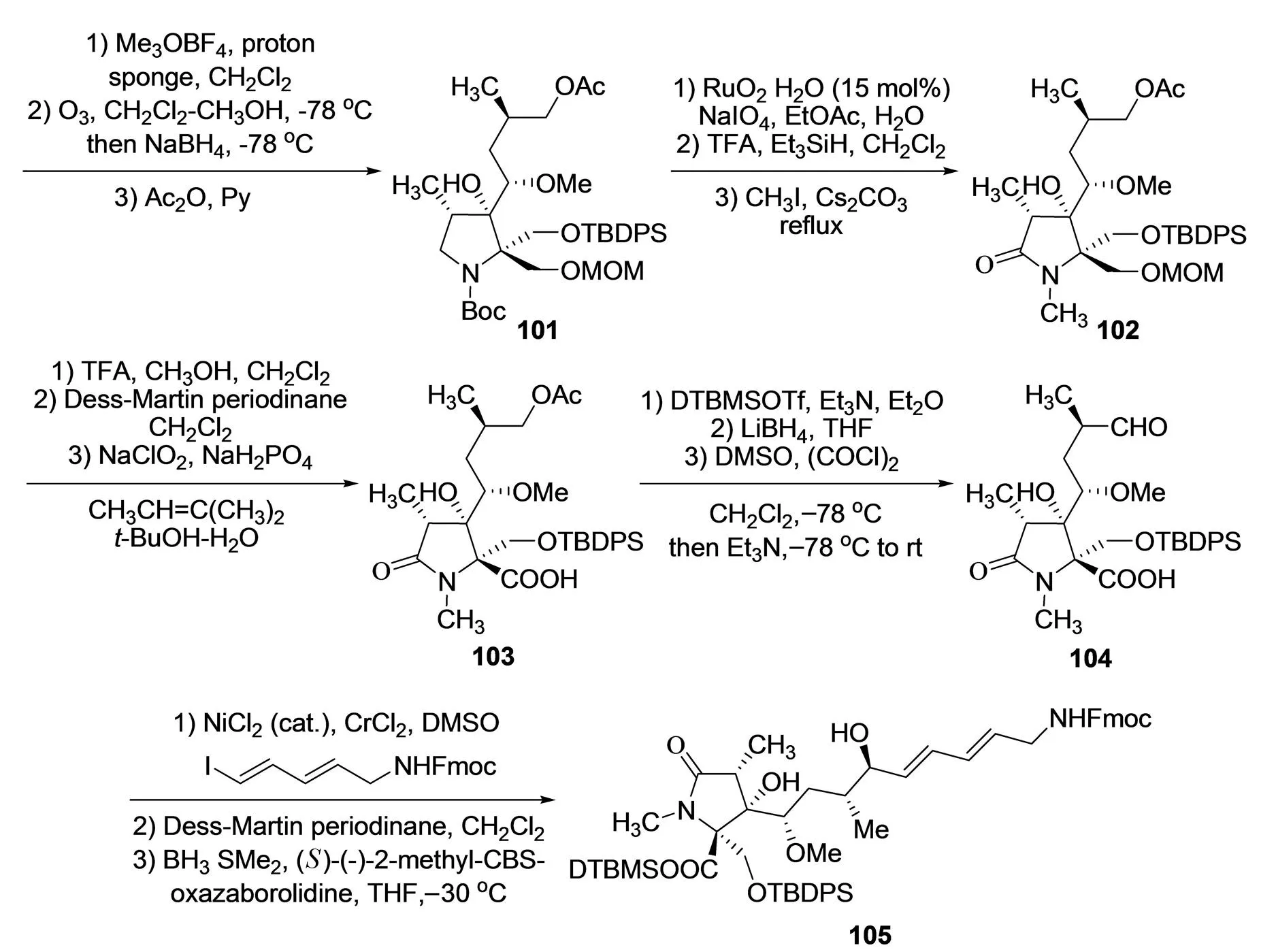
图12 DONOHOE等对噁唑霉素A的片段C的合成[31]Fig.12 DONOHOE′s synthesis of fragment C in oxazolomycin A[31]
2展望
噁唑霉素及其类似物的结构复杂性虽然给全合成工作带来了巨大挑战,但这类化合物具有广泛且独特的生物活性以及潜在临床实验的需求,已经引起了越来越多的有机化学家与药物化学家的关注. 根据这一领域的发展趋势以及对噁唑霉素及其类似物研究现状的了解,本文作者认为在今后相当长的时间内噁唑霉素及其类似物的全合成仍将是全合成化学研究的热点之一,主要研究将集中在引入新的方法学策略,设计全新的、高效的全合成路线,以及对已报道的合成路线的优化等方面.
参考文献:
[1] MORI T, TAKAHASHI K, KASHIWAHARA M, et al. Structure of oxazolomycin, a novelβ-lactone antibiotic [J]. Tetrahedron Lett, 1985, 26(8): 1073-1076.
[2] TAKAHASHI K, KAWABATA M, UEMURA D, et al. Structure of neooxazolomycin, an antitumor antibiotic [J]. Tetrahedron Lett, 1985, 26(8): 1077-1078.
[3] KANZAKI H, WADA K, NITODA T, et al. Novel bioactive oxazolomycin isomers produced byStreptomycesalbusJA3453 [J]. Biosci Biotechnol Biochem, 1998, 62(3): 438-442.
[4] RYU G, HWANG S, KIM S K. 16-Methyloxazolomycin, a new antimicrobial and cytotoxic substance produced by aStreptomycessp. [J]. J Antibiot, 1997, 50(12): 1064-1066.
[5] RYU G, KIM S K. Absolute stereochemistry determination of 16-methyloxazolomycin produced by aStreptomycessp. [J]. J Antibiot, 1999, 52(2): 193-197.
[6] OGURA M, NAKAYAMA H, FURIHATA K, et al. Structure of a new antibiotic curromycin a produced by a geneticality modified atrain ofStreptomyceshygroscopicus, a polyether antibiotic producing organism [J]. J Antibiot, 1985, 38(5): 669-673.
[7] OTANI T, YOSHIDA K I, KUBOTA H, et al. Novel triene-β-lactone antibiotics, oxazolomycin derivative and its isomer, produced byStreptomycessp. KSM-2690 [J]. J Antibiot, 2000, 53(12): 1397-1400.
[8] KENDE A S, KAWAMURA K, DEVITA R J. Enantioselective total synthesis of neooxazolomycin [J]. J Am Chem Soc, 1990, 112(10): 4070-4072.
[9] ONYANGO E O, TSURUMOTO J, IMAI N, et al. Total synthesis of neooxazolomycin [J]. Angew Chem Int Ed, 2007, 46(35): 6703-6705.
[10] ETO K, YOSHINO M, TAKAHASHI K, et al. Total synthesis of oxazolomycin A [J]. Org Lett, 2011, 13(19): 5398-5401.
[11] KENDE A S, KAWAMURA K, ORWAT M J. Enantioselective synthesis of theα,α-dimethyl-β-hydroxyacid subunit of the oxazolomycin antibiotics[J]. Tetrahedron Lett, 1989, 30(43): 5821-5824.
[12] WASMUTH D, ARIGONI D, SEEBACH D. Zum stereochemischen verlauf der biosynthese von 2-oxo-pantolacton: synthese von stereospezifisch indiziertem pantolacton aus äpfelsäure [J]. Helv Chim Acta, 1982, 65(1): 344-352.
[13] BULGER P G, MOLONEY M G, TRIPPIER P C. A multicomponent coupling strategy for the synthesis of the triene component of the oxazolomycin antibiotics [J]. Synlett, 2002, 1871-1873.
[14] BULGER P G, MOLONEY M G, TRIPPIER P C. A multicomponent coupling strategy suitable for the synthesis of the triene component of the oxazolomycin antibiotics [J]. Org Biomol Chem, 2003, 1(21): 3726-3737.
[15] WANG Z, MOLONEY M G. Synthesis of the middle fragment of oxazolomycin [J]. Tetrahedron Lett, 2002, 43(52): 9629-9632.
[16] ANDRUS M B, LEPORE S D, TURNER T M. Total synthesis of stipiamide and designed polyenes as new agents for the reversal of multidrug resistance [J]. J Am Chem Soc, 1997, 119(50): 12159-12169.
[17] ANDREWS M D, BREWSTER A G, MOLONEY M G. Highly functionalised pyroglutamates by intramolecular aldol reactions: Towards the pyroglutamate skeleton of oxazolomycin [J]. Synlett, 1997, 612-614.
[18] SEEBACH D, AEBI J D. α-Alkylation of serine with self-reproduction of the center of chirality [J]. Tetrahedron Lett, 1984, 25(24): 2545-2548.
[19] HEAVISIDE E A, MOLONEY M G, THOMPSON A L. Diastereoselective intramolecular aldol ring closures of threonine derivatives leading to densely functionalised pyroglutamates related to oxazolomycin [J]. RSC Adv, 2014, 4: 16233-16249.
[20] PAPILLON J P N, TAYLOR R J K. The first syntheses of the 1-oxo-2-oxa-5-azaspiro[3.4]octane ring system found in oxazolomycin [J]. Org Lett, 2000, 2(14): 1987-1990.
[21] CARLSEN P H G, KATSUKI T, MARTIN V S, et al. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds [J]. J Org Chem, 1981, 46(19): 3936-3938.
[22] MOHAPATRA D K, MONDAL D, GONNADE R G, et al. Synthesis of the spiro fusedβ-lactone-δ-lactam segment of oxazolomycin [J]. Tetrahedron Lett, 2006, 47(34): 6031-6035.
[23] CRIMMINS M T, KING B W, TABET E A, et al. Asymmetric aldol additions: use of titanium tetrachloride and (-)-sparteine for the soft enolization of N-acyl oxazolidinones, oxazolidinethiones, and thiazolidinethiones [J]. J Org Chem, 2001, 66(3): 894-902.
[24] EVANS D A, BARTROLI J, SHIH T L. Enantioselective aldol condensations. 2. erythro-selective chiral aldol condensations via boron enolates [J]. J Am Chem Soc, 1981, 103(8): 2127-2129.
[25] MITSUNOBU O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products [J]. Synthesis, 1981: 1-28.
[26] BENNETT N J, PRODGER J C, PATTENDEN G. A synthesis of a common intermediate to the lactone-pyrrolidinone ring systems in oxazolomycin A and neooxazolomycin [J]. Tetrahedron, 2007, 63(27): 6216-6231.
[27] XU J, LI X, WU J, et al. Synthesis of 5-alkyl-5-aryl-γ-lactams from 1-aryl-substituted nitroalkanes and methyl acrylate via Michael addition and reductive lactamization [J]. Tetrahedron, 2014, 70(25): 3839-3846.
[28] ZHANG X, SUN X, RAO W. TfOH-catalyzed tandem reactions of cyclopropyl alcohols with sulfonamides for the synthesis of pyrrolidines [J]. Chin J Org Chem, 2015, doi: 10.6023/cjoc201501036.
[29] YAMADA T, SAKAGUCHI K, SHINADA T, et al. Efcient asymmetric synthesis of the functionalized pyroglutamate core unit common to oxazolomycin and neooxazolomycin using Michael reaction of nucleophilic glycine Schiff base withα,β-disubstituted acrylate [J]. Tetrahedron: Asymmetry, 2008, 19(24): 2789-2795.
[30] MONDAL D, BERA S. A synthetic view of an analogue of the spiro-β-lactone-γ-lactam ring in oxazolomycins and lajollamycin [J]. Synthesis, 2010: 3301-3308.
[31] DONOHOE T J, O′RIORDAN T J C, PEIFER M, et al. Asymmetric synthesis of the fully elaborated pyrrolidinone core of oxazolomycin A [J]. Org Lett, 2012, 14(21): 5460-5463.
[责任编辑:任铁钢]
Progress in the synthesis of oxazolomycin and its analogs
LIANG Dawei1*, LAI Bate2, SU Jing3
(1.DepartmentofPharmacyandMedicalLaboratory,Ya'anVocationalCollege,Ya'an625000,Sichuan,China;
2.AstellasPharmaChina,Inc.,Shenyang110041,Liaoning,China; 3.NortheastPharmaceutical
Group,ShenyangNo.1PharmaceuticalCo.,Ltd.,Shenyang110141,Liaoning,China)
Abstract:Oxazolomycin and its relevant analogs are isolated from a strain of Streptomyces, all containing three fragments. Due to its structural originality and potent biological activities, these natural products have attracted considerable attention from the medicinal chemistry community. Since the first discovery, several research groups have reported the synthesis of oxazolomycin or its analogs. Here in, we detail the approaches taken by several research groups that are actively engaged in the synthesis of one or more of oxazolomycins based on the three fragments. The key synthetic strategies and methods of some reported routes are briefed. Moreover, suggestions are given on the development of the total synthesis of oxazolomycins.
Keywords:oxazolomycin; antibiotic; synthesis; research progress
作者简介:梁大伟 (1985-),男,讲师,研究方向为药物及药物中间体的合成.*通讯联系人,E-mail:ldawei@yahoo.com.
基金项目:雅安职业技术学院—药用分子研究创新团队项目(YZYTD-2015-01).
收稿日期:2015-05-28.
文章编号:1008-1011(2015)06-0643-10
中图分类号:O626.24
文献标志码:A
