叠氮桥对双核镍配合物磁性影响的密度泛函理论研究
2015-12-29边江鱼岳淑美张景萍
边江鱼 岳淑美 张 敏 张景萍
(1长春师范大学化学学院,长春130032;2东北师范大学化学学院,长春130024)
叠氮桥对双核镍配合物磁性影响的密度泛函理论研究
边江鱼1,*岳淑美1张 敏1张景萍2,*
(1长春师范大学化学学院,长春130032;2东北师范大学化学学院,长春130024)
结合对称性破损(BS)方法,采用不同的密度泛函理论(DFT)对反铁磁性μ-1,3-N3-Ni(II)叠氮配合物[LNi2(N3)](ClO4)2(L=pyrazolate)的磁特性进行了研究.结果显示,杂化密度泛函理论(HDFT)的计算结果与实验数据非常吻合,能够准确描述配合物的磁特性.磁轨道研究结果表明,配合物表现出较大的单占据轨道能量劈裂(0.93-0.99 eV),显示配合物的单占据轨道去简并化程度较大,且配合物中的2个磁通道(叠氮基、配体pyrazolate)中都分别存在有氮原子之间的p轨道重叠,这些都使得体系表现为反铁磁耦合作用.另外,配合物的磁性与叠氮桥和两金属离子间形成的二面角(τ,Ni-N-N-N-Ni)密切相关,τ从-55.38°逐渐变化到-1.5°的过程中,其反铁磁性逐渐增强,交换耦合常数(Jab)的绝对值逐渐增大,并在-11.95°处达到最大值(Jab=-151.02 cm-1).在此过程中,配合物中叠氮桥及其所连接的2个Ni离子与pyrazolate基配体L-中的2个桥原子N(4)、N(5)形成的七元环共平面性不断增强,即共平面性会诱导增强体系的反铁磁相互作用.
密度泛函理论-对称性破损方法;叠氮配合物;交换耦合常数;反铁磁相互作用
Received:January 15,2015;Revised:April15,2015;Published onWeb:April16,2015.
∗Corresponding authors.BIAN Jiang-Yu,Email:bianjy2002@163.com;Tel:+86-43186168210.
ZHANG Jing-Ping,Email:zhangjp162@nenu.edu.cn;Tel:+86-43185099372.
The projectwas supported by the Natural Science Foundation of Changchun NormalUniversity,China(2009-009,2010-030)and“Twelfth Five-Year Plan”Science and Technology Research Projectsof Jilin ProvincialDepartmentof Education,China(2011-192).
长春师范学院自然科学基金(长师院自科合字[2009]第009号,长师院自科合字政策[2010]第030号)和吉林省教育厅科学技术研究“十二五”规划项目(吉教科合字[2011]第192号)资助
©Editorialofficeof Acta Physico-Chimica Sinica
Key Wo rds:Density functionaltheory-broken symmetry(DFT-BS)method;Azido com p lex; Magnetic coupling constant;Anti-ferromagnetic interaction

图1 [LNi2(N3)]2+的结构Fig.1 Structureof[LNi2(N3)]2+Hydrogen atomsare notshown.
1 引言
分子基磁性材料是近几十年发展起来的新型材料,由于其结构和磁性质的多样性,受到了物理、化学和材料学家的广泛关注.1-5配合物分子的磁性除了与中心离子(高自旋载体)本身性质密切相关外,还取决于桥联配体的性质.通过选择适当的桥联配体,可以控制配合物的分子结构和磁性质.叠氮酸根(N3-)作为一种多功能的配体,不仅可形成多种不同的桥联方式和聚合结构,在磁性传递上也富有变化.利用丰富的桥联配位模式,叠氮离子和过渡金属离子可以构筑从零维到三维不同维度的聚合结构,4c,6-12在宏观上也表现出丰富的磁学行为,尤其是开关配合物的发现,9a引起了实验化学家和理论化学家极大的研究兴趣.
然而,面对形形色色的磁性材料,人们对其磁构效关系的研究还尚未成熟,因而采用理论手段来研究这些磁性体的磁构效关系非常必要.13,14由于双核过渡金属化合物结构简单,且具有代表性,在结构与磁性研究领域引起了广泛的重视.本文以反铁磁性μ-1,3-N3-Ni(II)叠氮配合物[LNi2(N3)](ClO4)2(L= pyrazolate)为研究对象,15运用密度泛函理论结合对称性破损(DFT-BS)方法对该配合物中的磁交换相互作用进行研究,采用不同密度泛函方法以及不同基组对标题金属配合物的交换耦合常数进行了计算,并分析叠氮桥与两金属离子间形成的二面角对配合物磁性的影响,从而揭示该磁性配合物的磁-结构相关问题,为实验研究提供重要的理论基础.
2 计算方法及模型
结合对称性破损(BS)方法,选取了几种密度泛函方法:广义梯度近似(GGA)(BP86,BPW 91,BLYP和PBE),局域自旋密度近似(LSDA)(SVWN, SVWN5)和杂化密度泛函理论(HDFT)(B3P86, B3LYP,B3PW 91和PBE0),采用LANL2DZ基组来计算配合物的交换耦合常数(Jab).为了测试基组效应对计算Jab的影响,选用B3PW 91和PBE0杂化密度泛函,计算了不同基组(LANL2DZ,6-31G,SDD和TZVP)下的Jab.全部计算工作采用Gaussian 03程序16完成.构建合适的对称性破损态(BS态)是计算磁交换耦合常数的关键,目前文献报道有多种方法可用来构建BS态的初始猜测,例如Yin等17通过自然键轨道(NBO)的定域化轨道来得到BS态的初始猜测.本文则通过Guess=m ix选项混合配合物的HOMO与LUMO轨道来破坏其轨道对称性,实现波函数的初始猜测.所有高自旋态(HS态)和BS态的波函数都通过Stable=opt选项进行了稳定性检测.
众所周知,Jab对分子结构参数非常敏感,键长和键角的微小变化都有可能引起Jab的明显变化,甚至可能引起磁学性质的转变,因此使用在气相中优化的几何结构计算得到的Jab通常并不能很好地重复实验结果,故本文中pyrazolate基双核Ni(II)μ-1,3-N3反铁磁性配合物[LNi2(N3)](ClO4)2(ClO4-为抗衡离子)中所有分子的原子坐标直接取自实验的X衍射单晶结构数据,15结构如图1所示.配合物中金属离子采用五配位模式:2个金属通过μ-1,3-N3相联,其他4个氮给体来自桥配体pyrazolate的侧面.
3 计算公式
两个磁中心的相互作用可用Heisenberg-Dirac van Vleck哈密顿模型来描述:

式中用Jab来表示两个磁中心a和b的未成对电子之间的交换耦合常数,它的符号和绝对值大小标志着相互作用的性质和强弱.Jab为正值,表示磁中心之间的相互作用性质为铁磁耦合,Jab为负值则表示磁中心之间是反铁磁耦合相互作用.S→a,S→b分别为磁中心a和b的总自旋角动量.
Jab可采用对称性破损方法根据以下公式计算得到:

其中YE(X)和Y<S2>分别表示采用X方法(UHF, UDFT等)计算的Y自旋态(高自旋态,低自旋态(LS))的总能量和自旋平方算符的平均值/期望值.
4 结果与讨论

表1 LANL2DZ基组下不同DFT方法计算所得配合物对称性破损态(BS)和五重态(QS)的总能量(E),自旋平方算符的平均值/期望值(S2)和交换耦合常数(Jab)Table 1 Totalenergies(E),expectation value of the totalspin angularmomentum squared(S2)for broken-symm etry(BS) and quintet(QS)states,and themagnetic coup ling constant(Jab)values for title com plex obtained by severalDFTmethodsw ith LANL2DZ basisset
4.1 交换耦合常数Jab
表1列出了不同密度泛函方法对配合物交换耦合常数的计算结果.从表1可以看出,尽管不同方法计算所得的Jab值均为负值,表明配合物的磁特性为反铁磁性,与实验观测结果一致,但Jab的计算值对不同的密度泛函方法也表现出了很大的依赖性.
从表1中可以清楚的看到,所有考察的DFT方法中,杂化密度泛函方法计算所得的Jab值最接近于实验值,而局域自旋密度近似方法计算所得的Jab值与实验结果偏差最大.采用杂化密度泛函方法(UB3PW 91,UB3LYP,UB3P86和UPBE0)计算得到的Jab值分别为-29.12,-38.38,-52.94和-44.23 cm-1,与实验观测值-(25.7±0.3)cm-1非常接近,尤其是UB3PW 91方法.而通过其他密度泛函和HF方法计算得到的Jab绝对值更大,与实验值偏差较大.计算所得Jab绝对值大小按如下顺序递增UB3PW 91<UB3LYP<UPBE0<UB3P86<UBPW 91<UBLYP<UBP86<UPBE<UHF<USVWN5<USVWN.
为了测试不同基组对配合物交换耦合常数计算的影响,选取UB3PW 91和UPBE0方法,在不同的基组(LANL2DZ,6-31G,SDD,TZVP)水平上对配合物的交换耦合常数进行了计算,结果列于表2.表2数据显示,采用UB3PW 91方法计算所得的Jab对基组的依赖性较大.其中TZVP基组得到的结果和实验值相差最大,SDD基组次之,而LANL2DZ和6-31G基组得到的Jab与实验值的吻合程度得到明显改进.对UPBE0方法来说,不同基组计算所得Jab相差不大,对基组依赖性较小.
4.2 分子磁轨道
在分子磁学中,定域在局域自旋中心的轨道称为局域磁轨道,高自旋态的单占据磁轨道(SOMOs)被看作分子磁轨道,通常,后者决定分子体系的磁行为.图2直观地描述了ROB3PW 91/LANL2DZ水平上对配合物计算所得的五重态的轨道.
根据Hoffmann理论,磁轨道和交换耦合常数之间的关系可以通过定性分析分子磁轨道来理解.从图2可以看出:该配合物磁轨道的主要贡献来自于Ni(II)的d轨道、叠氮和pyrazolate中桥联氮原子的p轨道.其中,HOMO-2和HOMO-3主要由Ni(II)的型轨道和pyrazolate中桥联氮原子π轨道组成,而HOMO和HOMO-1的主要贡献则来自于Ni(II)的dz2
型轨道和叠氮桥中氮原子间的π轨道,以及pyrazolate中桥联氮原子的π轨道.通过研究单占据轨道SOMOs的分布方式,可以看到HOMO-3中金属中心Ni(1)和pyrazolate中的N(4)、HOMO-2中金属中心Ni(2)和pyrazolate中的N(5)分别存在p-d轨道重叠.HOMO-1和HOMO单占据分子轨道中叠氮的氮原子之间存在有p轨道重叠,从而有利于配合物的反铁磁相互作用.叠氮桥(μ-1,3-N3)和pyrazolate中桥联氮原子(N(4),N(5))分别是配合物自旋中心之间的磁耦合相互作用传递的两条通道,且两条通道对体系反铁磁耦合相互作用都有贡献.
表3列出了不同HDFT方法计算得到的配合物单占据轨道能量(E)及单占据轨道能量劈裂值(ΔE=结果显示,不同方法得到的单占据轨道能量劈裂值基本一致,约0.93-0.99 eV,表明配合物的单占据轨道去简并化程度较大,是体系表现为反铁磁耦合作用的原因.

表2 不同基组计算所得配合物对称性破损态和五重态的总能量,自旋平方算符的平均值/期望值和交换耦合常数Table 2 Totalenergies,spin angularm omentum for broken-symmetry and quintetstatesand themagnetic coup ling constant values for title comp lex obtained w ith severalbasissets
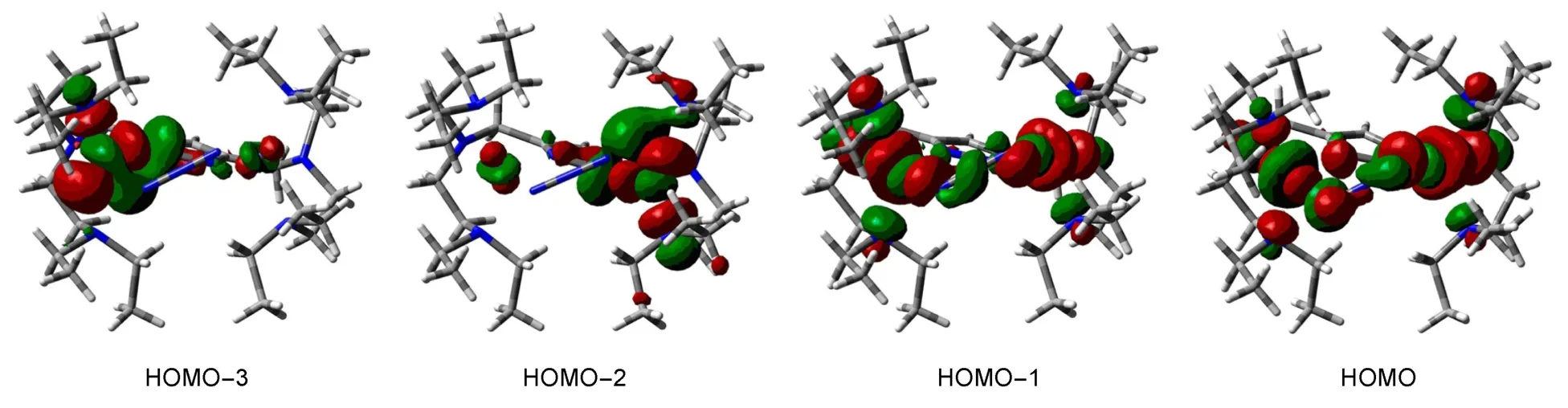
图2 所得配合物在ROB3PW 91/LANL 2DZ水平上计算所得的五重态的SOMOsFig.2 Singly occupied molecular orbitals(SOMOs)for the title com p lex in the quintet state at the ROB3PW 91/LANL2DZ level

表3 HDFT对配合物计算所得单占据轨道能量(E)及单占据轨道能量劈裂值(ΔE)Table 3 Energiesof SOMOs(E)and energy sp litting between SOMOs(ΔE)for the title comp lex obtained by hybrid density functional theory(HDFT)methods
4.3 自旋密度分析
研究模型结构的自旋布居分布,有助于理解磁中心的磁交换耦合行为.根据分子轨道理论,自旋离域可以解释为未成对电子由磁中心向配体原子转移,1而自旋极化则主要来自于Pauli原理导致的电子交换作用,18并在相继连接的配体原子上产生交替符号的自旋分布.自旋离域和自旋极化机理曾成功用于处理单核和同双核体系.19据文献20报道,铁磁高自旋态和反铁磁低自旋态时体系自旋密度分布对定性的理解体系自旋相关性有着非常重要的指导作用.
根据Mulliken布居分析,采用不同的杂化密度泛函方法对配合物低自旋态时部分原子的自旋密度分布进行了计算,结果列于表4,其中正负号分别表示α和β自旋.结果表明,不同密度泛函方法计算所得体系各原子自旋密度的符号是一致的,也就是说,所有方法对自旋离域和自旋极化效应的测评是一致的,只是程度不同.正是因为程度的不同导致不同方法计算所得的交换耦合常数有很大的差异.我们选取Jab计算值与实验值吻合最好的B3PW 91方法的计算结果来讨论.
由图3可见,金属磁中心Ni(1)和Ni(2)的自旋分布相反,分别为1.630和-1.619,与磁中心相连的配体pyrazolate上的氮原子N(4)和N(5)和其相邻金属中心的自旋密度符号相同(分别为+0.060和-0.084),说明存在有从金属中心到配体的自旋离域效应.连接两个金属中心的叠氮基中,两个终端氮原子的自旋密度符号相反,分别为(N(1)-0.082, N(3)0.079),而叠氮基的中心氮原子上的自旋密度(0.002)因两终端氮原子(N(1)和N(3))的反向自旋极化作用被大大的抑制而明显接近于零.21,22整体来看,从一个金属中心通过叠氮桥到另一个金属中心这一路径上,自旋密度正负交替分布,另外,配体中N(4)和N(5)的自旋密度符号也不同,这些都体现了配合物的自旋极化效应.因此,在该反铁磁性配合物中,自旋离域效应和自旋极化效应对体系的磁交换相互作用都有贡献,体系的反铁磁性相互作用是自旋离域和自旋极化协同作用的结果.

表4 不同DFT方法计算所得配合物低自旋态部分原子的自旋密度分布Table 4 Spin density distributions for partatom sof the title com plex in the singlet stateobtained by severalDFTmethods

图3 [LNi2(N3)]2+部分原子自旋密度分布图Fig.3 Spindensitydistributionsforpartatomsof[LNi2(N3)]2+

图4 Ni-N-N-N-Ni二面角(τ)对Jab计算值的影响Fig.4 Ni-N-N-N-Nidihedralangle(τ) dependence for Jabvalues
4.4 Ni-N-N-N-Ni二面角τ对磁性的影响
在该配合物中,对叠氮基和pyrazolate基配体L-两个反铁磁耦合通道来说,构象效应对交换相互作用有着重要的影响.然而配体L-在双核Ni(II)配合物[LNi2(N3)]2+中的空间结构限制了pyrazolate基配体上氮原子的有效变化.因此本文在上述研究的基础上采用UB3PW 91/LANL2DZ方法研究了该反铁磁性配合物中的Ni-N-N-N-Ni二面角τ的变化对配合物磁性质的影响.
在该配合物的晶体结构中观察到Ni-N-N-N-Ni二面角为-55.38°,以此为出发点逐渐将之变化到9°,其他的结构保持不变.计算体系的交换耦合常数,并将Jab随τ变化的趋势示于图4(计算所得数据见Supporting Information).
从图4可以看到,在该二面角变化区域中,该磁性配合物的Jab保持负值,也就是说两个磁中心之间的相互作用一直为反铁磁相互作用.随着τ的增加, [LNi2(N3)]2+的反铁磁相互作用先增强后减弱.当τ的变化在(-17.09°--1.5°)区间时,Jab的绝对值较大,并在-11.95°处达到最大值(Jab=-151.02 cm-1).在交换耦合常数不断增大的过程中,配合物中Ni-N-NN-Ni与pyrazolate基配体L-中的两个桥原子N(4)、N (5)形成的七元环,其共平面性随τ的增大而不断增强,即共平面性会诱导体系反铁磁相互作用增强,其原因应源于共平面性有利于磁通道中N原子之间的轨道重叠.Jab随τ的变化遵循下面方程关系:

相关系数为0.9908.以上所得结论和Leibeling等9a合成的双核Ni(II)-azide双稳态磁性配合物性质基本一致.Ni(II)-azide配合物在低温时τ=4.34°,表现为较强的反铁磁性(Jab=-(81.0±1.5)cm-1),高温时τ=-46.46°,表现为弱反铁磁性(Jab=-(24.0±1.0)cm-1).
5 结论
通过DFT-BS方法对反铁磁性[LNi2(N3)](ClO4)2配合物的磁性质进行了研究.结果显示,杂化密度泛函理论(HDFT)计算所得的交换耦合常数与实验值很好地吻合,能够准确描述配合物的磁特性.磁轨道分析表明,配合物大的单占据轨道的能量劈裂,是其表现为反铁磁耦合作用的主要原因.该配合物中存在两条磁通道,分别为叠氮基和配体pyrazolate.两条磁通道中氮原子之间都存在p轨道重叠,对体系反铁磁耦合相互作用都有贡献.从自旋布居分布角度来看,该配合物中反铁磁性相互作用是自旋离域和自旋极化协同作用的结果.另外,配合物中Ni-N-N-N-Ni与pyrazolate基配体L-中的两个桥原子N(4)、N(5)形成的七元环的共平面性越强,体系的反铁磁相互作用越强,即共平面性会诱导增强体系的反铁磁相互作用.
Suppo rting In fo rm ation:available free of charge via the internetathttp://www.whxb.pku.edu.cn.
(1)Kahn,O.MolecularMagnetism;VCH Publications:New York, 1993.
(2)(a)Carroll,R.L.;Gorman,C.B.Angew.Chem.Int.Edit.2002, 41,4378.doi:10.1002/1521-3773(20021202)41:23<4378::AIDANIE4378>3.0.CO;2-A (b)Bousseksou,A.;Molnár,G.;Matouzenko,G.Eur.J.Inorg. Chem.2004,2004,4353. (c)Zhang,P.;Zhang,L.;Tang,J.K.Dalton Trans.2015,44, 3923. (d)Antonis,N.A.;Zacharias,G.F.;Madhu,M.J.Phys.: Condes.Matter2015,27,052202.
(3)(a)Umezono,Y.;Fujita,W.;Awaga,K.J.Am.Chem.Soc.2006, 128,1084.doi:10.1021/ja057207i (b)Jeannin,O.;Clérac,R.;Fourm igué,M.J.Am.Chem.Soc. 2006,128,14649. (c)Bréfuel,N.;Shova,S.;Tuchagues,J.P.Eur.J.Inorg.Chem. 2007,2007,4326. (d)Koner,R.;Hazra,S.;Fleck,M.;Jana,A.;Lucas,C.R.; Mohanta,S.Eur.J.Inorg.Chem.2009,2009,4982.
(4)(a)Delferro,M.;Graiff,C.;Marchiò,L.;Elviri,L.;Mazzani, M.;Riccò,M.;Predieri,G.Eur.J.Inorg.Chem.2011,2011, 3327.doi:10.1002/ejic.201100385 (b)Cardona-Serra,S.;Clemente-Juan,J.M.;Coronado,E.; Gaita-A riño,A.;Suaud,N.;Svoboda,O.;Bastardis,R.; Guihéry,N.;Palacios,J.J.Chem.-Eur.J.2015,21,763. (c)Zhang,Y.Q.;Luo,C.L.Int.J.Quantum Chem.2006,106, 1551.
(5)(a)Frecus,B.;Oprea,C.I.;Panait,P.;Ferbinteanu,M.; Cimpoesu,F.;Gîrţu,M.A.Theor.Chem.Acc.2014,133, 1470.doi:10.1007/s00214-014-1470-0 (b)Guedes,G.P.;Florencio,A.S.;Carneiro,J.W.M.;Vaz,M. G.F.Solid State Sci.2013,18,10. (c)Triki,S.;Gómez-García,C.J.;Ruiz,E.;Sala-Pala,J.Inorg. Chem.2005,44,5501. (d)Jia,L.H.;Liu,A.C.;Mu,Z.E.;Chen,Y.F.Acta Phys.-Chim.Sin.2011,27,1595.[贾丽慧,刘安昌,牟宗娥,陈云峰.物理化学学报,2011,27,1595].doi:10.3866/PKU. WHXB20110736 (e)James,M.;Brant,C.Inorg.Chim.Acta 2012,384,189.
(6)(a)Feng,P.L.;Stephenson,C.J.;Am jad,A.;Ogawa,G.;Barco, E.D.;Hendrickson,D.N.Inorg.Chem.2010,49,1304.doi: 10.1021/ic902298y (b)M ilios,C.J.;Inglis,R.;Vinslava,A.;Prescimone,A.; Parsons,S.;Perlepes,S.P.;Christou,G.;Brechin,E.K.Chem. Commun.2007,26,2738. (c)Sun,H.L.;Wang,Z.M.;Gao,S.Chem.-Eur.J.2009,15, 1757. (d)Gu,Z.G.;Song,Y.;Zuo,J.L.;You,X.Z.Inorg.Chem. 2007,46,9522. (e)Liu,T.F.;Fu,D.;Gao,S.;Zhang,Y.Z.;Sun,H.L.;Su,G.; Liu,Y.J.J.Am.Chem.Soc.2003,125,13976.
(7)(a)Sasmal,S.;Hazra,S.;Kundu,P.;Majumder,S.;A liaga-A lcalde,N.;Ruiz,E.;Mohanta,S.Inorg.Chem.2010,49, 9517.doi:10.1021/ic101209m (b)Demeshko,S.;Leibeling,G.;Dechert,S.;Meyer,F.Dalton Trans.2006,28,3458. (c)Mukherjee,P.S.;Maji,T.K.;Escuer,A.;Vicente,R.;Ribas, J.;Rosair,G.;Mautner,F.A.;Chaudhuri,N.R.Eur.J.Inorg. Chem.2002,2002,943.
(8)(a)M ilios,C.J.;Prescimone,A.;Sanchez-Benitez,J.;Parsons, S.;Murrie,M.;Brechin,E.K.Inorg.Chem.2006,45,7053.doi:10.1021/ic061035o (b)Tandon,S.S.;Bunge,S.D.;Sanchiz,J.;Thompson,L.K. Inorg.Chem.2012,51,3270.
(9)(a)Leibeling,G.;Demeshko,S.;Dechert,S.;Meyer,F.Angew. Chem.Int.Edit.2005,44,7111. (b)Demeshko,S.;Leibeling,G.;Maringgele,W.;Meyer,F.; Mennerich,C.;K lauss,H.H.;Pritzkow,H.Inorg.Chem.2005, 44,519.
(10)(a)Papaefstathiou,G.S.;Escuer,A.;Vicente,R.;Font-Bardia, M.;Solans,X.;Perlepes,S.P.Chem.Commun.2001,23,2414. (b)Meyer,F.;Kircher,P.;Pritzkow,H.Chem.Commun.2003, 6,774. (c)Zhang,X.M.;Wang,Y.Q.;Song,Y.;Gao,E.Q.Inorg. Chem.2011,50,7284. (d)Brunet,G.;Habib,F.;Cook,C.;Pathmalingam,T.;Loiseau, F.;Korobkov,I.;Burchell,T.J.;Beauchemin,A.M.;Murugesu, M.Chem.Commun.2012,48,1287. (e)Sengupta,O.;Mukherjee,P.S.Inorg.Chem.2010,49,8583. (f)Lin,S.Y.;Zhao,L.;Guo,Y.N.;Zhang,P.;Guo,Y.;Tang,J. K.Inorg.Chem.2012,51,10522.
(11)Chakraborty,A.;Rao,L.S.;Manna,A.K.;Pati,S.K.;Ribas,J.; Maji,T.K.Dalton Trans.2013,42,10707.doi:10.1039/ c3dt32526a
(12)(a)Mukherjee,S.;Mukherjee,P.S.Dalton Trans.2013,42, 4019.doi:10.1039/c2dt32802j (b)Mukherjee,S.;Mukherjee,P.S.AccountsChem.Res.2013, 46,2556. (c)Mukherjee,S.;Mukherjee,P.S.Cryst.Growth Des.2014, 14,4177.
(13)(a)Bian,J.Y.;Chang,Y.F.;Zhang,J.P.J.Phys.Chem.A 2008, 112,3186.doi:10.1021/jp711121z (b)Noh,E.A.A.;Zhang,J.P.Chem.Phys.2006,330,82. (c)Noh,E.A.A.;Zhang,J.P.Theochem 2009,896,54. (d)Noh,E.A.A.;Zhang,J.P.Theochem 2008,867,33.
(14)(a)Clarke,C.S.;Jornet-Somoza,J.;Mota,F.;Novoa,J.J.; Deumal,M.J.Am.Chem.Soc.2010,132,17817.doi:10.1021/ ja1057746 (b)Onofrio,N.;Mouesca,J.M.Inorg.Chem.2011,50,5577. (c)Sasmal,S.;Hazra,S.;Kundu,P.;Dutta,S.;Rajaraman,G.; Sañudo,E.C.;Mohanta,S.Inorg.Chem.2011,50,7257. (d)Biswas,R.;Mukherjee,S.;Kar,P.;Ghosh,A.Inorg.Chem. 2012,51,8150. (e)Pramanik,K.;Malpaharia,P.;Mota,A.J.;Colacio,E.;Das, B.;Lloret,F.;Chandra,S.K.Inorg.Chem.2013,52,3995.
(15)Leibeling,G.;Demeshko,S.;Bauer-Siebenlist,B.;Mayer,F.; Pritzkow,H.Eur.J.Inorg.Chem.2004,2004,2413.
(16)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.etal.Gaussian 03, Revision C.01;Gaussian Inc.:Pittsburgh,PA,2003.
(17)(a)Yin,B.;Li,J.L.;Bai,H.C.;Wen,Z.Y.;Jiang,Z.Y.;Huang, Y.H.Phys.Chem.Chem.Phys.2012,14,1121.doi:10.1039/ C1CP22928A (b)Yu,Y.;Li,C.;Yin,B.;Li,L.J.;Huang,Y.H.;Wen,Z.Y.; Jiang,Z.Y.J.Chem.Phys.2013,139,054305.
(18)Ruiz,E.;Cirera,J.;Alvarez,S.Coord.Chem.Rev.2005,249, 2649.doi:10.1016/j.ccr.2005.04.010
(19)Cano,J.;Ruiz,E.;A lvarez,S.;Verdaguer,M.Comments Inorg. Chem.1998,20,27.doi:10.1080/02603599808032749
(20)M itani,M.;Mori,H.;Takano,Y.;Yamaki,D.;Yoshioka,Y.; Yamaguchi,K.J.Chem.Phys.2000,113,4035.doi:10.1063/ 1.1286418
(21)(a)Willet,R.D.;Gatteschi,D.;Kahn,O.Magneto-Structural Correlations in Exchange Coupled Systems;Reidel:Dordrecht, 1985. (b)O'Connor,C.J.Research Frontiers in Magnetochemistry; World Scientific:Singapore,1993. (c)Chen,C.T.;Suslick,K.S.Coord.Chem.Rev.1993,128, 293.
(22)(a)Koner,R.;Lin,H.H.;Wei,H.H.;Mohanta,S.Inorg.Chem. 2005,44,3524.doi:10.1021/ic048196h (b)Nanda,K.K.;Thompson,L.K.;Bridson,J.N.;Nag,K. J.Chem.Soc.Chem.Commun.1994,11,1337. (c)Arriortua,M.I.;Cortés,R.;Mesa,J.L.;Lezama,L.;Rojo, T.;Villeneuve,G.Transition Met.Chem.1988,13,371.
Effec ts o f Azido Bridge on Magnetic Properties o f Dinuc lear Nickel Com p lexes:Density Func tional Theo ry Stud ies
BIAN Jiang-Yu1,*YUE Shu-Mei1ZHANGMin1ZHANG Jing-Ping2,*
(1DepartmentofChemistry,Changchun NormalUniversity,Changchun 130032,P.R.China;2DepartmentofChemistry,NortheastNormalUniversity,Changchun 130024,P.R.China)
Themagnetic properties of the antiferromagnetic comp lexμ-1,3-N3-Ni(II)[LNi2(N3)](ClO4)2(L= pyrazolate)were investigated using density functional theory(DFT)calculations combined w ith the broken symmetry approach.The calculation results obtained using the hybrid density functional theory(HDFT)agree wellw ith the experimentaldata,and accurately describe themagnetic properties of com plex.The large energy splitting,0.93-0.99 eV,between singly occupiedmolecularorbitals indicates that there is strong non-degeneracy between them,and the two coup ling paths(azido and pyrazolate)in the com p lex show that there is overlap between the p orbitals of the N atoms.All these factors contribute to the antiferromagnetism of the com plex. The magnetic properties of the com p lex are also closely related to the dihedralangleτof Ni-N-N-N-Ni.The antiferromagnetism of the com p lex increases asτdecreases from-55.38°to-1.5°;them aximum absolute value o fmagnetic coup ling constant(Jab)occurs at-11.95°(Jab=-151.02 cm-1).During this process,the coplanarity of the seven-membered ring,which consists of two Ni(II),one azido,and two bridging nitrogen atoms (N(4)and N(5)),is enhanced,i.e.,cop lanarity increases the antiferromagnetism of the com plex.
O641
icle]
10.3866/PKU.WHXB201504162 www.whxb.pku.edu.cn