复方马度米星铵预混剂中尼卡巴嗪含量测定方法的研究
2015-10-13刘素梅杜红鸽方忠意张跃京臧和英邱天宝
刘素梅,杜红鸽,方忠意,张跃京,臧和英,邱天宝,王 丽
(河南省兽药饲料监察所,河南 郑州 450008)
复方马度米星铵预混剂中尼卡巴嗪含量测定方法的研究
刘素梅,杜红鸽,方忠意,张跃京,臧和英,邱天宝,王丽
(河南省兽药饲料监察所,河南 郑州 450008)
建立了高效液相色谱法测定复方马度米星铵预混剂中尼卡巴嗪的含量,采用Waters X-Bridge C18(150 NN 4.6 mm,5 μm)色谱柱,乙腈-水(用磷酸调pH至3.0)为流动相,梯度洗脱,流速为1.0 ml/min,检测波长0~5 min为297 nm,5 min开始为350 nm,柱温30℃,进样体积为20 μl。HDP的回归方程为:y=23452x-456,相关系数r=0.99999,DNC的回归方程为:y=102388x+5296,相关系数r=0.9999,即HDP在0.29~29.12 μg/ml范围内具有良好的线性,DNC在0.71~70.90 μg/ml范围内具有良好的线性。该方法简便、准确、灵敏度高、重复性好,可以通过同时测定尼卡巴嗪中HDP和DNC来计算尼卡巴嗪的含量。
复方马度米星铵预混剂;尼卡巴嗪;HDP;DNC;高效液相色谱法
复方马度米星铵预混剂收载于农业部公告第1435号[1]中,为尼卡巴嗪和马度米星铵的复方制剂,作为饲料添加剂用于防治鸡球虫病。方中尼卡巴嗪为4,4′-二硝基均二苯脲(DNC)和2-羟基-4,6-二甲基嘧啶(HDP)的复合物,对鸡的多种艾美耳球虫,如柔嫩、脆弱、毒害巨型、堆形、布氏艾美耳球虫均有良好的防治效果[2]。
标准中尼卡巴嗪含量测定采用的紫外分光光度法,并且通过只测定4,4′-二硝基苯脲(DNC)的含量来控制尼卡巴嗪的含量,样品需经反应显色,操作较烦琐,且样品溶液不稳定,重现性差,每次显色后必须快速测定,无法同时测定多批样品。只有沈祥广等[3]通过高效液相色谱法对HDP和DNC进行了同时测定,但该方法流动相配制烦琐,且HDP易和溶剂峰重合在一起。根据兽药典委员会的要求,将尼卡巴嗪原料及制剂中尼卡巴嗪含量测定方法进一步完善,修订为高效液相色谱法,并拟将该标准收载在《中国兽药典》2015年版中。
1 仪器与试药
Waters Alliance高效液相色谱系统:包括2695分离系统、2489紫外检测器、Empower2色谱工作站;METTLER XP205电子天平;MILIPORE超纯水处理器等。
复方马度米星铵预混剂由濮阳泓天威药业有限公司(批号为GR201307002)、河南孟成生物药业股份有限公司(批号为20130701)和浙江汇能动物药品有限公司(批号为413022518)提供;尼卡巴嗪对照品由中国兽医药品监察所提供,批号为H0330903;液相用甲醇、乙腈为色谱纯试剂,水为超纯水,其他试剂为分析纯。
2 方法与结果
2.1溶液的制备
2.1.1对照品溶液的制备精密称取尼卡巴嗪对照品50 mg,置50 ml量瓶中,加二甲基亚砜20 ml,超声使溶解,加乙腈稀释至刻度,摇匀;临用前用乙腈-水(6∶4)稀释成浓度为1、5、10、40、80和100 μg/ml的系列对照品溶液,过0.45 μm微孔滤膜即得。
2.1.2供试品溶液的制备取本品0.63 g,精密称定,置50 ml量瓶中,加二甲基亚砜20 ml,超声提取20 min,加乙腈稀释至刻度,摇匀;精密量取上清液2 ml,置50 ml量瓶中,加乙腈-水(6∶4)稀释至刻度,摇匀。
2.2色谱条件的选择
尼卡巴嗪中HDP具有较大的极性,在反相色谱柱上保留较弱,很容易和溶剂二甲基亚砜重合在一起,参考不同文献[4-7],选取C18柱为色谱柱,考察不同的溶剂系统,最终采用乙腈-水(用磷酸调节pH值至3.0i 0.1)系统[8]。考虑到HDP保留较弱,出峰快,DNC出峰慢,且预混剂所用辅料为玉米芯,空白辅料有吸收峰,为避免这些吸收峰对组分的影响,选用了梯度洗脱,具体程序见表1。

表1 梯度程序表
经光电二极管阵列检测器检测,HDP的最大吸收波长为297 nm,DNC最大吸收波长为350 nm,所以在检测过程中需要切换波长,0~5 min为297 nm,5 min开始为350 nm。
2.3线性范围
精密称取尼卡巴嗪对照品50 mg,置50 ml量瓶中,加二甲基亚砜20 ml,超声溶解,加乙腈定容至刻度,摇匀;用乙腈-水(6∶4)稀释成浓度为1、5、10、40、80和100μg/ml的系列对照品溶液,过0.45 μm微孔滤膜,注入液相色谱仪,记录色谱图,以对照品溶液浓度与相应的组分峰面积算回归方程和相关系数,HDP的回归方程为:y=23452x-456,相关系数r=0.99999,DNC的回归方程为:y=102388x+ 5296,相关系数r=0.9999,即HDP在0.29~29.12 μg/ml范围内具有良好的线性,DNC在0.71~70.90 μg/ml范围内具有良好的线性。
2.4准确度
取空白辅料0.63 g,共取9份,精密称定,分别加入尼卡巴嗪原料40 mg、50 mg和60 mg共3个浓度水平,每个水平3份。按照2.1.2项下供试品制备方法进行处理,进高效液相色谱仪分析,按外标法以峰面积计算,结果见表2。
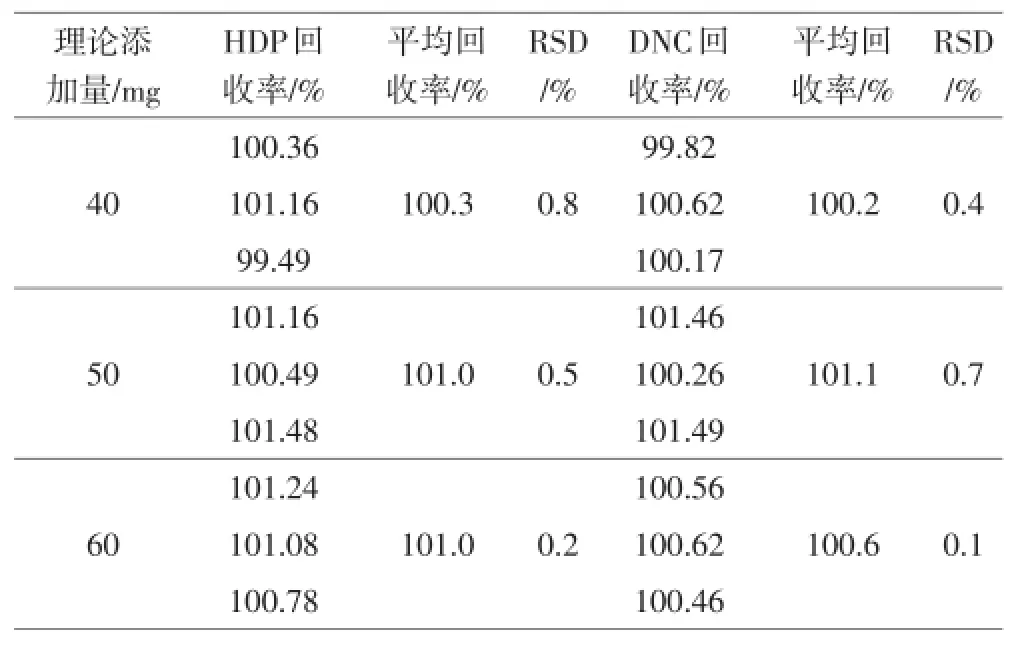
表2 复方马度米星铵预混剂准确度试验结果
从结果可以看出,采用高效液相色谱法同时测定两个组分的含量,回收率高。
2.5精密度
取同一批次样品0.63 g,共取6份,分别精密称定,按2.1.2项下供试品制备方法进行处理,进高效液相色谱仪分析,按外标法以峰面积计算含量,HDP的RSD为0.8%,DNC的RSD为0.4%,结果表明,该方法具有很好的精密度。
2.6专属性
经过考察生产企业的复方马度米星铵预混剂生产工艺,在生产过程中使用的可能影响到药物成分质量的试剂有苯甲醇和乙酸乙酯,笔者考察了这两种试剂对尼卡巴嗪色谱峰的影响。称取空白辅料0.63 g,分别加入1ml的苯甲醇和乙酸乙酯及2 mg的马度米星铵,按2.1.2项下供试品制备方法进行处理,进高效液相色谱仪分析,在该色谱条件下尼卡巴嗪两主峰无干扰。
2.7稳定性
取供试品0.63 g,按2.1.2项下供试品溶液制备方法进行处理,分别在0、1、8、12和24 h进样,考察供试品溶液稳定性,两组分峰面积RSD分别为0.8%和0.5%,结果显示,样品在24 h内稳定。
2.8耐用性
用XBridge C18(5 μm,4.6 ON 150 mm)色谱柱分别考察了不同柱温、不同流速和流动相不同pH值对色谱峰的影响,结果发现,不同柱温和不同流速对色谱峰的柱效、分离度等影响不大。流动相的水相pH值为2.5和3.5时,两主峰的理论塔板数下降幅度很大,pH值为3.5时,DNC峰展宽,出现肩峰;pH值为2.8和3.2时,两主峰的理论塔板数分别较pH值为2.5和3.5提高很多,能达到标准需求,所以pH值应尽量控制在3.0i 0.2。
2.93批样品测定
取3批复方马度米星铵预混剂,每批3份,分别精密称定,按2.1.2项下供试品溶液制备方法进行处理,进高效液相色谱仪分析,按外标法以峰面积计算含量及RSD,结果见表3。

表3 三批样品测定结果
由表3可知,该方法重复性好,可以实现HDP和DNC的同时测定。
3 讨论与结论
原标准中尼卡巴嗪含量测定采用的紫外分光光度法,并且通过只测定4,4′-二硝基苯脲(DNC)的含量来控制尼卡巴嗪的含量,供试品经反应显色,操作较烦琐,且样品不稳定,重现性差。修改后采用高效液相色谱法,两种组分同时测定,结果以两组分含量的和来计算,操作较原来简便了好多,样品的重现性很好,较原来的方法有很大的优越性。
HDP保留较弱,DNC保留很强,出峰慢,且预混剂所用辅料为玉米芯,空白辅料有吸收峰,为避免这些吸收峰对组分的影响,选用了梯度洗脱。
该方法简便、快速、重复性好,可以进行大批量样品的同时测定,对尼卡巴嗪中的两个组分进行控制,可以更好地控制制剂的质量。
[1]农业部兽药评审中心.农业部公告第1435号[S].2010.
[2]中国兽药典委员会.兽药使用指南化学药品卷[S].2011.
[3]沈祥广,贺利民,余静贤,等.尼卡巴嗪原料药及制剂中4,4′-二硝基均苯二脲与2-羟基-4,6-二甲基嘧啶的HPLC法同时测定[J].分析测试学报.2010,29(5):523-526.
[4]邵庆均,张金枝,宣士荣,等.反相高效液相色谱法测定饲料中尼卡巴嗪含量[J].分析化学.2003,31(11):1376-1379.
[5]陈丽,谭宝玲,冯建文.HPLC法检测饲料预混剂中尼卡巴嗪的含量[J].饲料工业.2006,27(14):52-53.
[6]孙雷,张骊,刘智宏,等.鸡蛋和鸡肉中尼卡巴嗪残留检测高效液相色谱-串联质谱法研究[J].中国兽药杂志. 2008,42(5):1-4.
[7]周伟伟,钟平,薛伟芳,等.鸡组织中尼卡巴嗪残留HPLC检测方法的研究[J].中国兽医学报.2009,29(12):1590-1592.
[8]刘素梅,班付国,韩立,等.高效液相色谱法测定尼卡巴嗪含量的方法建立[J].中国兽药杂志.2014,48(5):51-55.
S859.79
1004-5090(2015)11-0005-03
2015-08-21)
