噻吩在双金属有机多孔材料Ni- Cu/BTC上吸附性能的研究
2015-08-30王亭亭浙江师范大学化学与生命科学学院浙江金华3004浙江师范大学地理与环境科学学院浙江金华3004
何 乔,王亭亭,马 娜,代 伟*(.浙江师范大学化学与生命科学学院,浙江 金华 3004;.浙江师范大学地理与环境科学学院,浙江 金华 3004)
噻吩在双金属有机多孔材料Ni- Cu/BTC上吸附性能的研究
何乔1,王亭亭1,马娜2,代伟1*(1.浙江师范大学化学与生命科学学院,浙江 金华 321004;2.浙江师范大学地理与环境科学学院,浙江 金华 321004)
噻吩类硫化物的脱除是燃油能否实现生产超低硫清洁燃油的关键.本文研究了双金属有机多孔材料 Ni-Cu/BTC吸附燃油中噻吩的性能.结果表明,Ni-Cu/BTC对模型油中苯并噻吩的吸附动力学过程满足拟二级动力学模型.Langmuir、Freundlich和Dubinin-Radushkevich (D-R)3种等温吸附模型均可较好地描述Ni-Cu/BTC对苯并噻吩的等温吸附行为(Freundlich>D-R>Langmuir).热力学参数表明Ni-Cu/BTC对噻吩的吸附是自发的吸热过程.
Ni-Cu/BTC;噻吩;吸附
燃油中含硫化合物燃烧后产生的 SOx使发动机尾气净化器中的催化剂中毒,影响催化转化器的性能发挥,导致CO、烃类和 NOx等污染物排放量增多,造成严重的空气污染.随着世界各国对环境质量重视程度的提高,燃油中的硫含量受到了严格的限制.美国已经执行了汽油和柴油硫含量分别为30µg/g和15µg/g的立法标准[1],欧盟规定燃油中的硫含量小于 10µg/g[2].目前工业燃油二次精制的基本技术是加氢精制.反应总压为3~8MPa,反应温度 573~633K[3],工艺条件比较苛刻.尽管如此,如果将噻吩类硫化物完全脱除,实现低硫或无硫清洁燃油,需将加氢反应器加大至少七倍,氢气消耗和能耗也随之大幅增加,并且导致油品质量的下降. 因此,研发新型材料和技术脱除燃油中噻吩类硫化物是实现超低硫清洁燃油的关键.
通过吸附活性位和硫化物之间特殊的相互作用,吸附法能选择性地捕获苯并噻吩类硫化物,实现燃油深度净化.吸附法的关键是基底吸附剂的研发.金属有机多孔材料(Metal-Organic Porous Materials,MOMs)是由金属离子和有机配体通过配位键联接而成的有机-无机杂化材料.由于其多变的结构,高的比表面积和有序的孔结构特点,近几十年来在多孔材料研究领域倍受关注.以往的MOMs材料大多是单金属和单配体间的一对一配位合成方式,最近文献报道了含有双金属不饱和配位的双金属有机多孔材料.由于双金属不饱和配位具有组合、共同、协同效应,使其在吸附应用领域有广阔的应用前景.
MOMs表面的不饱和Ni、Cu金属离子与噻吩中的硫原子形成π络合物,噻吩硫原子中的π电子进入金属离子的4s轨道形成σ健,金属离子3d轨道的电子又会进入到噻吩的反π轨道,形成d-π*反健.σ健和d-π*健的形成产生了络合作用.产物是Ni和Cu离子与噻吩形成的π络合物.噻吩硫化物和非硫芳香烃化合物都能通过电子与金属发生两种作用:一是噻吩硫原子和一个金属作用形成的η1S键,二是噻吩硫原子和两个金属作用形成的S-μ3键[1].双金属有机多孔材料在染料废水净化等领域已经有一定的研究进展,然而在深度脱除燃油中噻吩类硫化物仍需要深入研究[4-7].另外,MOMs应用在燃油深度研究大多集中在动态法,即通过测定吸附穿透曲线来计算吸附剂的穿透和饱和容量.对于静态法的研究工作较少,因此,本文采用自制的Ni-Cu/BTC为脱硫剂,研究了噻吩在 Ni-Cu/BTC材料上的吸附等温线和吸附动力学性能,以期为利用MOMs材料深度脱除燃油中噻吩类硫化物提供理论及技术支撑.
1 材料与方法
1.1Ni-Cu/BTC制备及表征
采用本课题组自制的 Ni-Cu/BTC[8].具体合成步骤:将1g的Cu(NO3)2.3H2O(Sigma Aldrich公司)、1g的 Ni(NO3)2.6H2O 和 1g对苯二甲酸(H2BTC,Sigma Aldrich公司)在强烈搅拌下溶于50mL的N,N-二甲基甲酰胺(DMF,Sigma Aldrich公司)中.待溶解之后,将溶液置于自升压反应釜中358K恒温20h.通过固液分离得到蓝色产品,将样品浸渍在无水乙醇 72h.最后,将样品在真空氛围下加热到373K恒温12h,即得到Ni-Cu/BTC样品.用美国麦克公司ASAP 2020物理吸附仪进行N2吸脱附分析.先将样品在真空473K下处理6h,然后在 77K下测定其对 N2的吸脱附等温线.DFT方法计算得到的孔径分布图显示孔径集中在1~2nm之间.制备的Ni/Cu-BTC样品的孔容为0.43cm3/g,BET比表面积为1190m2/g.
1.2模型油配制及硫含量分析
按一定质量比例分别配成噻吩与正辛烷的模型油将噻吩(硫)含量稀释至 2000μg/g(760μg/g)备用.用气相色谱(GC7890,火焰光度检测器FPD)分析样品管中噻吩的含量.柱箱温度 333K,分流比40/1,汽化室温度493K,氢焰温度493K,EC-5毛细管柱(L=30m; i,d=0.30μm):进样体积为1μL:采用外标法定量测定苯并噻吩浓度.
1.3吸附实验
移取25mL的模型油放入100mL锥形瓶中,然后放入一定量的金属有机多孔材料 Ni-Cu/BTC,将混合液置于恒温振荡器中以200r/min的振荡强度振荡至预先设定的时间,反应结束后采用离心分离的方式进行固液分离.分别采用气相色谱FPD检测器测定上清液中噻吩的浓度.根据(1)式计算出平衡吸附量q.

式中:q为平衡吸附量,mg/g:C0为模型油初始苯并噻吩含量,mg/L;Ce为吸附平衡时的苯并噻吩含量,mg/L;V为模型油能够用量,mL:m为Ni-Cu/ BTC用量,mg.
2 结果与讨论
2.1扫描电镜能谱元素分析
样品Ni/Cu-BTC在合成过程中,由于空间位阻等原因,金属离子除了与对苯二甲酸配体配位以外,还会结合一些小的溶剂分子来满足其配位数的要求,如水、DMF等.当合成的样品Ni/Cu-BTC在真空氛围下加热一段时间后,这些小分子就会从骨架中排出,金属离子的配位就成不饱和状态,骨架结构仍然稳定,这意味着 Ni/Cu-BTC材料具有发生π络合吸附的吸附位,具有不饱和金属配位的多孔材料亦称为 π-络合吸附剂[1,2].扫描样品的制备:将样品在超声波中分散,将分散后样品沉积到载波片上,然后进行喷金.扫描电子显微镜(SEM)仪器为菲利普公司生产,型号为XL30ESEM 环境扫描电子显微镜,操作电压为30KV,测定时为低真空(<20,torr). Ni/Cu-BTC的扫描电镜能谱图及各元素组成结果如图1和表1所示.由表1可知,Ni/Cu-BTC中含有23.5wt%的Ni,26.1wt%的Cu,证明此吸附剂具有双金属特征,此结果符合相关文献报道[1,8].

图1 Ni-Cu/BTC扫描电镜能谱Fig.1 EDS analysis of Ni-Cu/BTC
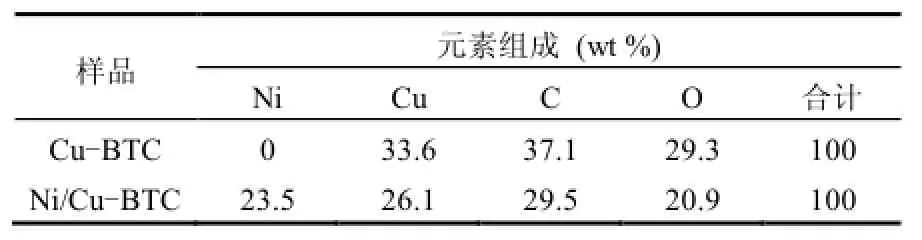
表1 扫描电镜能谱各元素组成Table 1 Element composition of EDS analysis
2.2吸附动力学
图2和图3分别为不同初始噻吩浓度和不同吸附温度条件下双金属有机多孔材料 Ni-Cu/ BTC对模型油中苯并噻吩的吸附动力学曲线.由图可知,Ni-Cu/BTC对噻吩的单位吸附量随着吸附时间的增加而增加,直至达到吸附平衡.在相同的吸附时间条件下,Ni-Cu/BTC对噻吩的吸附量随模型油中噻吩初始浓度的升高而增加.当模型油中的噻吩浓度越高,可供双金属有机多材料Ni-Cu/BTC吸附的噻吩就越多,同时模型油主体噻吩浓度与Ni-Cu/BTC外表面液膜的膜内噻吩浓度之间的浓度差越大,噻吩向Ni-Cu/BTC吸附剂表面迁移的动力也就越大.因此,增大模型油中噻吩的初始浓度有利于提高双金属有机多孔材料 Ni-Cu/BTC的噻吩吸附量.此外,在相同的吸附时间条件下,双金属有机多孔材料Ni-Cu/ BTC对噻吩的吸附量随温度的增加而增大,说明温度越高越有利于Ni-Cu/BTC对噻吩的吸附,也说明了该吸附过程是吸热反应.当温度升高时,会使模型油中噻吩克服Ni-Cu/BTC表面液膜阻力的能力增强[9],而有利于Ni-Cu/BTC表面吸附的噻吩向内部孔道迁移,导致Ni-Cu/BTC对苯并噻吩的吸附量增加.
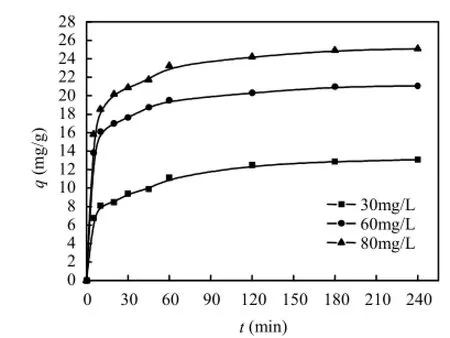
图2 不同初始浓度下Ni-Cu/BTC对噻吩的吸附动力学Fig.2 Adsorption kinetics of thiophene on Ni-Cu/BTC at various initial concentrations
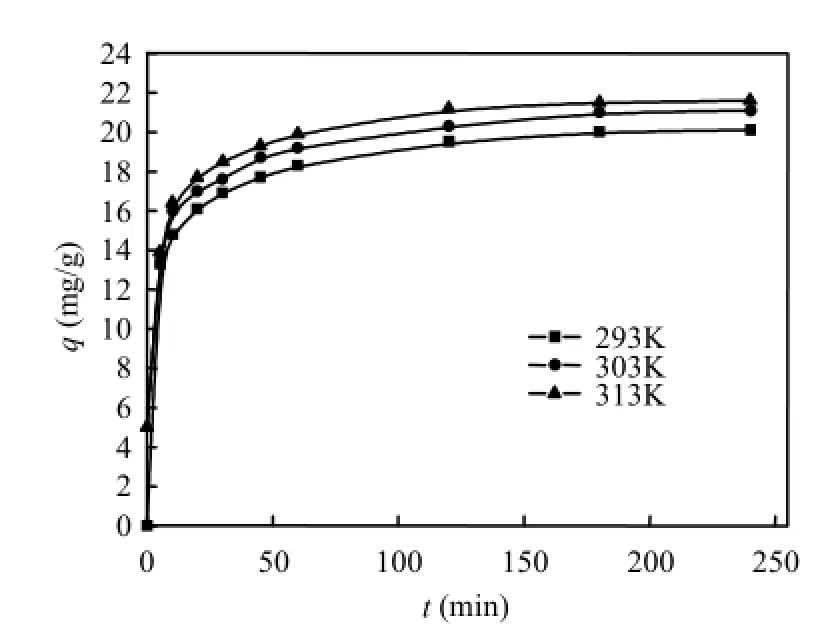
图3 不同温度条件下Ni-Cu/BTC对噻吩的吸附动力学Fig.3 Adsorption kinetics of thiophene on Ni-Cu/BTC at various temperatures
采用准一级和准二级动力学吸附模型对图1和图 2的实验数据进行拟合.准一级动力学模型的数学方程可表达为[10-11]:


式中:k1为准一级动力学速率常数,1/min:t为反应时间,min:qt和qe分别为t时刻和平衡时刻吸附剂对吸附质的单位吸附量,mg/g.准二级动力学模型的数学方程可表达为[12]:式中:k2为准二级动力学速率常数,g/mg·min.分别以ln(qe-qt)对t和t/qt对t作图,由直线的斜率和截距可以求出动力学的理论平衡吸附容量qe,cal和速率常数k1、k2的值,qe,exp为静态吸附平衡时,吸附剂实验条件下所测得浓度变化,并通过公式(1)计算的饱和吸附量,结果见表2和表3.由表2和表3可见,准二级动力学模型比准一级动力学更适合用于描述Ni-Cu/BTC对苯并噻吩的吸附过程.
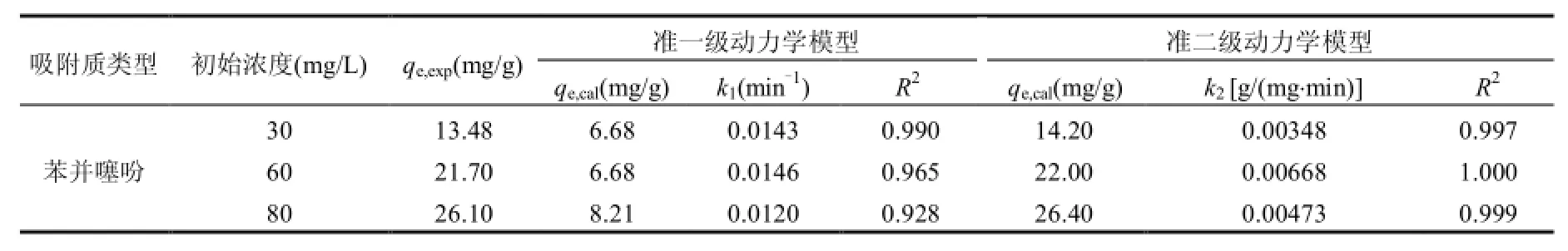
表2 不同初始浓度条件下Ni-Cu/BTC吸附噻吩的动力学参数Table 2 Kinetic models rate constants for benzothiophene adsorption onto MOF-5at various initial concentrations

表3 不同反应温度条件下Ni-Cu/BTC吸附噻吩的动力学参数Table 3 Kinetic models rate constants for thiophene adsorption onto Ni-Cu/BTC at various temperature
准一级和准二级动力学模型不能识别扩散机制的作用.因此,本研究将进一步采用颗粒内扩散模型识别扩散机制是否为Ni-Cu/BTC吸附噻吩过程的速率限制步骤.颗粒内扩散模型的数学方程可表达为[12]:

式中:ki为颗粒内扩散速率常数,mg/(g·min0.5);C为截距.根据该模型,如果吸附过程涉及颗粒内扩散机制,则qt对t0.5的曲线应该是线性的:如果颗粒内扩散机制是吸附过程的唯一速率限制步骤,则 qt对t0.5直线应通过原点:如果qt对t0.5直线不通过原点,则颗粒内扩散机制不是吸附过程唯一的速率限制步骤[15].图4为Ni-Cu/BTC吸附噻吩的qt对t0.5曲线.由图4可见,Ni-Cu/BTC对噻吩的吸附过程可以分为三个阶段:(i)第一阶段为快速的外表面吸附阶段:(ii)第二阶段为逐渐吸附阶段,颗粒内扩散是该阶段吸附过程的速率限制步骤:(iii)第三阶段为最终平衡阶段.第二阶段的拟合曲线不通过原点,这说明颗粒内扩散不是逐渐吸附阶段唯一的速率限制步骤,逐渐吸附阶段的速率限制步骤既包括液膜扩散也包括颗粒内扩散.
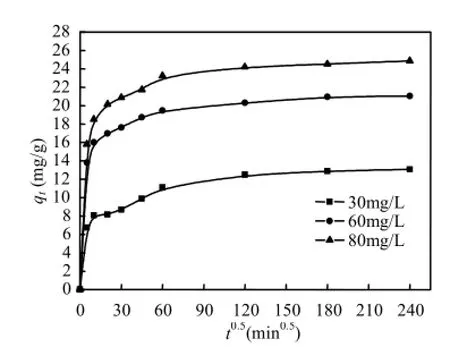
图4 qt对t0.5曲线Fig.4 Plots of qtversus t0.5
采用阿伦尼乌斯公式对Ni-Cu/BTC吸附噻吩过程的表观吸附活化能进行了计算.阿伦尼乌斯公式可表述为[13]:

式中:Ea为表观吸附活化能,kJ/mol:T为反应温度,(K):A为指前因子:R为理想气体常数,8.314J/ (mol·K).以1/T 为横坐标和ln(k2)为纵总坐标作图,由斜率确定Ni-Cu/BTC吸附噻吩过程的表观活化能为6.65kJ/mol.有研究指出,如果扩散机制是吸附过程的速率限制步骤,则吸附活化能值应小于25~30kJ/mol[13].根据Ea值的计算结果,证明扩散机制是Ni-Cu/BTC吸附噻吩过程的速率限制步骤.
2.3吸附等温线
图5为Ni-Cu/BTC对噻吩的等温吸附线.由图可知,不同吸附温度条件下 Ni-Cu/BTC对噻吩的平衡吸附量随着模型油中噻吩平衡浓度的增加而增加.此外,Ni-Cu/BTC对噻吩的平衡吸附量随着吸附温度的增加而增大,这说明吸附温度越高越有利于Ni-Cu/BTC对噻吩的吸附行为.采用 Langmuir、Freundlich和 Dubinin-Radushkevich (D-R)等温吸附模型对图 4的实验数据进行拟合.Langmuir等温吸附模型的数学方程可表述为[14]:

式中:qe为Ni-Cu/BTC对噻吩的平衡单位吸附量,mg/g:qm为噻吩在 Ni-Cu/BTC上的最大吸附容量,mg/g:Ce为模型油中苯并噻吩的平衡浓度,mg/L:KL为Langmuir常数(L/mg).为进一步分析Langmuir等温吸附模型,引入一个无量纲的常数-分离因数(RL).RL可通过公式(7)进行计算[14]:

式中:C0为模型油中噻吩的初始浓度,mg/L.根据RL的数值大小,可将Langmuir等温吸附模型的吸附类型分为四类:(i)0<RL<1,优惠吸附:(ii) RL> 1,非优惠吸附:(iii) RL= 1,线性吸附:(iv) RL= 0,不可逆吸附[14].Freundlich等温吸附模型的数学方程可表述为[14]:

式中:KF为与吸附容量有关的Freundlich常数:1/n为与吸附强度有关的经验系数.1/n介于0.1和1之间说明吸附过程属于优惠吸附[9].D-R等温吸附模型的数学方程可表述为[14]:

式中:q0为 Ni-Cu/BTC对噻吩的最大吸附容量,mol/g:KD为与吸附能有关的常数,mol2/kJ2:ε为Polanyi吸附势,ε = RTln(1+1/Ce):Ce为模型油中吸附质的平衡浓度,mol/L.平均吸附自由能E(kJ/mol)是指单位数量的噻吩从模型油中无穷远处移动到固体表面时自由能的变化,可通过公式(10)进行计算[14]:
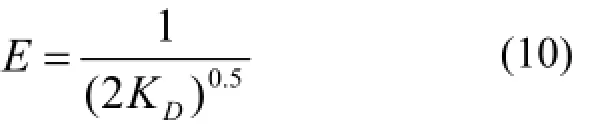
E的数值可以用来判别吸附反应的类型:(i)1<E<8kJ/mol,吸附为物理吸附:(ii)8<E<16kJ/mol,吸附以化学吸附为主[14].

图5 Ni-Cu/BTC上噻吩的吸附等温线Fig.5 Adsorption isotherms of thiophene on Ni-Cu/BTC
分别以Ce/qe对Ce、ln(qe)对ln(Ce)和ln(qe)对 ε2作图,由直线的斜率和截距可以求出Langmuir、Freundlich和D-R等温吸附模型的参数,结果见表3.由表3可知,Langmuir、Freundlich 和 D-R等温吸附模型均可以很好地描述Ni-Cu/BTC对噻吩的吸附.根据Langmuir等温吸附模型计算得到的当吸附温度为 303K条件下Ni-Cu/BTC对噻吩的最大单位吸附量分别为25.28mg/g.计算得到的RL值均大于0小于1,1/n值均大于0.1小于1,这说明了Ni-Cu/BTC对噻吩的吸附是优惠吸附.根据D-R等温吸附模型计算得到的Ni-Cu/BTC吸附噻吩的平均自由能为16~18kJ/mol,这说明 Ni-Cu/BTC吸附噻吩的过程是一个以化学吸附为主的过程,Ni-Cu/BTC结构中的不饱和镍和铜离子与噻吩发生 π络合吸附作用,这符合相关文献报道[18].根据相关文献报道[1-2]运用惰性气体(如氦气)保护,高温(673K左右)吹扫,再用有机溶剂(如乙醇)洗涤的方法,吸附饱和的络合吸附剂Ni-Cu/BTC可以实现再生,再生容量大于90%.
2.4热力学参数
为了确定 Ni-Cu/BTC对噻吩的吸附过程是否为自发过程以及是否为吸热或放热过程,本研究对 Ni-Cu/BTC吸附噻吩的热力学参数进行了计算.热力学参数包括吉布斯自由能变化(ΔG0)、焓变(ΔH0)和熵变(ΔS0)的计算公式可表述为[15]:
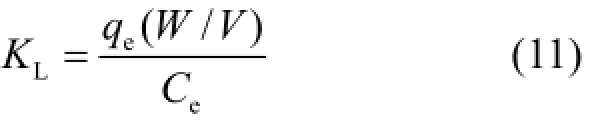

式中:KL为化学反应平衡常数,L/mol; V是溶液体积,L;W,吸附剂质量,g.根据公式(11)、(12)和(13)计算得到的Ni-Cu/BTC吸附噻吩的热力学参数值见表5.由表5可知,当反应温度为293、303和313K时,计算得到的ΔG0为负值,这说明Ni-Cu/BTC吸附噻吩的过程是热力学自发过程.由表5还可以发现计算得到的ΔH0为正值,这说明Ni-Cu/BTC吸附噻吩的过程是吸热过程.由表5还可以发现计算得到的ΔS0为正值,这说Ni-Cu/BTC吸附噻吩增加了固-液界面上物质的无序程度.
表4 Ni-Cu/BTC吸附噻吩的等温吸附模型参数Table 4 Isotherm model parameters for thiophene adsorption onto Ni-Cu/BTC

表5 Ni-Cu/BTC吸附噻吩的热力学参数Table 5 Thermodynamic parameters for thiophene sorption onto Ni-Cu/BTC
3 结论
Ni-Cu/BTC对噻吩具有很好的吸附能力,吸附过程遵循准二级动力学模型.Langmuir、Freundlich和 D-R模型可以很好地描述噻吩在Ni-Cu/BTC上的等温吸附行为.双金属有机多孔材料Ni-Cu/BTC吸附噻吩的过程是自发吸热过程.含有双金属不饱和为的Ni-Cu/BTC在燃油深度脱硫领域具有良好的应用潜力.
[1] Yang R T,Herna´ndez-Maldonado A J,Yang F H. Desulfurization of transportation fuels with zeolites under ambient conditions [J]. Science,2003,301(1):79-81.
[2] Wei Dai,Jue Hu,Limei Zhou,et al. He Huang. Removal of dibenzothiophene with composite adsorbent MOF-5/Cu(I) [J]. Enery and Fuels,2013,27(2):816-821.
[3] 林世雄主编,石油炼制工程 [M]. 北京:石油工业出版社,2000:153-160.
[4] G. Blanco-Brieva,J.M. Campos-martin,S.M. Al-zahrani,J.L.G. Fierro. Removal of Refractory organic sulfur compounds in fossil fuels using MOF sorbents [J]. Global Nest Journal,2010,12(3):296-304.
[5] Cychosz K A,Wong-Foy A G,Matzger A J. Liquid phase adsorption by microporous coordination polymers: removal of organosulfur compounds [J]. J. Am. Chem. Soc,2008,130(22):6938-6939.
[6] Liu Baojian,Zhu Yinbang,Liu Shiwang,et al. Adsorption equilibrium of thiophenic sulfur compounds on the Cu-BTC metal-organic framework [J]. J. Chem. Eng. Data,2012,57(4):1326-1330.
[7] Khan N A,Jun J W,Jeong J H,et al. Remarkable adsorption performance of a metal-organic framework,vanadium-benzenedicarboxylate (MIL-47),for benzothiophene[J]. Chem. Commun.,2011,47(4):1306-1308.
[8] Hu Jue,Yu Huijing,Dai Wei,et al. Enhanced adsorptive removal of hazardous anionic dye “congo red” by a Ni/Cu mixedcomponent metal-organic porous material [J]. RSC Adv.,2014,4(66):35124-35130.
[9] 刘焱,王世和,吴玲琳.工业废渣基复合除磷材料的吸附动力学及热力学分析 [J]. 东南大学学报:自然科学版,2009,39(6):1231-1235.
[10] 董庆洁,周学永,邵仕香.锆、铁水合氧化物对磷酸根的吸附 [J].离子交换与吸附,2006,22(4):363-368.
[11] Huang H M,Xiao X M,Yan B. Ammonium removal from aqueous solutions by using natural Chinese (Chende) zeolite as adsorbent [J]. Journal of Hazardous Materials,2010,175(1-3):247-252.
[12] Mezenner N Y,Bensmaili A. Kinetics and thermodynamic study of phosphate adsorption on iron hydroxide-eggshell waste [J]. Chemical Engineering Journal,2009,147(2/3):87-96.
[13] Katal R,Baei M S,Rahmati H T,et al. Kinetic,isotherm and thermodynamic study of nitrate adsorption from aqueous solution using modified rice husk [J]. Journal of Industrial and Engineering Chemistry,2012,18(1):295-302.
[14] Zhao Y F,Zhang B,Zhang X,et al. Preparation of highly ordered cubic NaA zeolite from halloysite mineral for adsorption of ammonium ions [J]. Journal of Hazardous Materials,2010,178(1-3):658-664.
[15] Wei Dai,Yaping Zhou,Shengnan Li,et al. Thiophene capture with complex adsorbent SBA-15/Cu(I) [J]. Ind. Eng. Chem. Res.,2006,45(23):7892-7896.
Adsorptive performance of thiophene by bimetallic organic porous material Ni-Cu/BTC.
HE Qiao1,WANG Ting-ting1,MA Na2,DAI Wei1*(1College of Chemistry and Life Sciences,2College of Geography and Environmental Sciences,Zhejiang Normal University,Jinhua 321004,Zhe Jiang Province,China).
China Environmental Science,2015,35(7):1983~1989
Removal of thiophenic compounds from fuels plays a key role in production of ultra low sulfur clean fuel. The adsorptive performance of thiophene with bimetallic organic porous material Ni-Cu/BTC was investigated using batch experiments. Results showed that the adsorption kinetics data of thiophene onto Ni-Cu/BTC from model fuel could be well described by a pseudo-second-order model. The adsorption equilibrium data of thiophene onto Ni-Cu/BTC fitted well to the Langmuir,Freundlich and Dubinin-Radushkevich (D-R) isotherm models (Freundlich>D-R>Langmuir). Thermodynamic parameters showed that the adsorption of thiophene onto Ni-Cu/BTC was spontaneous and endothermic in nature.
Ni-Cu/BTC;thiophene;adsorption
X511
A
1000-6923(2015)07-1983-07
2014-12-15
浙江省2014年新苗人才计划项目(2014R404055),浙江省公益性技术应用研究计划项目(2013C31094)
* 责任作者,副教授,daiwei@zjnu.cn
代伟(1975-),男,辽宁本溪人,副教授,博士,主要从事新型材料制备及其吸附性能研究.发表论文50余篇.
