高效液相色谱法测定左卡尼汀注射液的含量及杂质A
2015-08-19袁彩君朱天新
袁彩君+++朱天新


[摘要] 目的 建立一种以阴离子色谱柱为分离介质测定左卡尼汀注射液含量及杂质A的高效液相色谱法。 方法 色谱柱为PhenoSphere 5u SAX 80A(250 mm×4.60 mm),柱温30℃;流动相为乙腈∶磷酸盐溶液(磷酸二氢钾6.81 g/L,以氢氧化钠溶液调节pH为4.7)=65∶35;检测波长为205 nm,进样量20 μl;流速1.0 ml/min,外标法计算。 结果 左卡尼汀和杂质A得到有效分离,分离度>1.5,辅料及流动相不干扰测定;左卡尼汀的检出限和定量限分别为0.5、2 μg/ml,左卡尼汀杂质A的检出限、定量限分别为20、50 ng/ml;左卡尼汀在1034~8274 μg/ml范围内线性关系良好(r2=0.99996),杂质A在0.25~25 μg/ml范围内线性关系良好(r2=0.99987);左卡尼汀和杂质A平均回收率分别为101.29%(RSD=0.55%)、100.35%(RSD=1.79%)。 结论 该方法专属、准确、简便,可用于左卡尼汀注射液的含量和杂质A测定。
[关键词] 左卡尼汀注射液;左卡尼汀;杂质A;高效液相色谱法;测定
[中图分类号] R927.2 [文献标识码] A [文章编号] 1674-4721(2015)07(a)-0008-06
Content and impurity A of levocarnitine injection determined by high performance liquid chromatography
YUAN Cai-jun1 ZHU Tian-xin2
1.Department of Pharmacy,Zhumadian Central Hospital in Henan Province,Zhumadian 463000,China;2.Xintai Pharmaceutical Co., LTD. of Henan Province,Zhumadian 463000,China
[Abstract] Objective To establish a high performance liquid chromatography (HPLC) method determining the content and impurity A of levocarnitine injection selecting anion chromatographic column as separation medium. Methods The column was PhenoSphere 5u SAX 80A (250×4.60 mm) and the column temperature was 30 ℃.The mobile phase was the ratio of acetonitrile and phosphate solution (potassium dihydrogen phosphate of 6.81 g/L regulated by sodium hydroxide solution with the pH of 4.7) was 65:35.The detection wavelength was 205 nm and injection volume was 20 μl.The flow velocity was 1.0 ml/min and the outcome were calculated by external standard method. Results Levocarnitine and impurity A was effectively separated,and the degree of separation was over 1.5.Determination was not interfered by adjuvant material or mobile phase.The limit of detection (LOD) and limit of quantitation (LOQ) of levocarnitine was 0.5 μg/ml and 2 μg/ml respectively.The LOD and LOQ of impurity A in levocarnitine was 20 ng/ml and 50 ng/ml respectively.Ranging from 1034 to 8274 μg/ml,levocarnitine displayed a good linear relationship (r2=0.99996).Ranging from 0.25 to 25 μg/ml,impurity A displayed a good linear relationship (r2=0.99987).The average recovery rate of levocarnitine and impurity A was 101.29% (RSD=0.55%) and 100.35% (RSD=1.79%). Conclusion The established method is specific,accurate and convenient,which can be used for the determination of content and impurity A in levocarnitine injection.
[Key words] Levocarnitine injection;Levocarnitine;Impurity A;High performance liquid chromatography;Determination
左卡尼汀(levocarnitine,LC)又称左旋肉毒碱,其主要功能是将长链脂肪酸运至线粒体进行β-氧化,并产生能量;它还能加速乙酰乙酸的氧化,在酮体代谢中起到积极作用。LC参与异亮氨酸和亮氨酸等代谢产物的转运,有益于氨基酸的正常代谢,并参与精子的成熟过程;其对脂溶性维生素A、E和D及钙、磷的吸收还有协同作用[1]。
LC注射液首先由意大利Sigma-Tau 公司研制生产,并于1999 年12月通过FDA批准上市,用于尿毒症、心血管疾病如心绞痛[2-3]、心肌梗死[4]、心力衰竭等[5]、中枢神经系统退行性疾病[6]、肾病[7-9]、肝病[10-11]、糖尿病[12]等疾病的治疗,临床应用越来越广泛。LC与LC制剂的测定方法较多,有酶法[13]、放射性同位素法[14]、光学法[15]、滴定法[16]、柱前衍生化HPLC法[17-19]、反相色谱法[20-22]等,其中酶法、同位素法、光学法、滴定法精密度、重复性较差;柱前衍生化HPLC法主要用于血药、尿液浓度监测[17-18]或异构体测定[19],尚无检测杂质A的报道。国家药品标准[22]收载的方法不能很好地将LC和其杂质A分开、欧盟药典[23]所选用的色谱柱为丙胺基甲基硅烷键合硅胶柱(aminopropylm ethylsilyl silicagel),在国内尚未找到相应的色谱柱,且该标准对LC与其杂质A的分离度限度要求(≥0.9)较低。本试验建立了以阴离子交换柱为分析介质的LC注射液高效液相色谱法(HPLC),可用于该注射液的含量测定及杂质A控制。
1 仪器与试药
高效液相色谱仪(岛津LC-10A二元泵),电子分析天平(北京赛多利斯仪器系统有限公司),水为注射用水,乙腈为色谱纯,磷酸二氢钾、氢氧化钠均为分析纯;左卡尼汀注射液,河南欣泰药业有限公司生产,规格:1 g∶5 ml,批号20100801、20100802、20100803;左卡尼汀注射液,意大利Sigma-Tau生产,规格:1g∶5 ml,批号080480;LC对照品(美国药典会,批号CAT. No.1359903),LC杂质A对照品:美国药典会,批号CAT. No.1359925。
2 方法与结果
2.1 色谱条件
色谱柱:PhenoSphere 5u SAX 80A(250 mm×4.60 mm)检测波长:205 nm,柱温:30℃。流动相:乙腈∶磷酸盐(磷酸二氢钾6.81 g/L,以氢氧化钠溶液调节pH为4.7)=65∶35。流速为1.0 ml/min,进样量为20 μl。
2.2 溶液制备
2.2.1 LC对照品溶液 精密称取适量LC对照品,置25 ml 容量瓶中,加流动相溶解并稀释成约5 mg/ml 溶液,振摇、混匀备用。
2.2.2 参比溶液a 精密称取LC杂质A对照品适量用水溶解并稀释成每毫升含0.25 mg的溶液,再精密量取1.0 ml用流动相稀释至10.0 ml,使成25 μg/ml的溶液(杂质A浓度为LC对照品溶液浓度的0.5%),振摇、混匀备用。
2.2.3 参比溶液b 精密称取LC杂质A对照品适量用水溶解并稀释成每毫升含1.0 mg的溶液,再精密量取1.0 ml用流动相稀释至10.0 ml,使成100 μg/ml的溶液,振摇、混匀备用。
2.2.4 参比溶液c 精密称取LC对照品适量,用参比溶液b溶解并稀释成每毫升约含LC对照品10 mg的溶液,振摇、混匀备用。
2.2.5 供试品溶液 精密量取取LC注射液1.25 ml,用流动相稀释至50 ml,振摇、混匀备用。
2.3 专属性
2.3.1 破坏性试验 ①未破坏:取LC约0.1 g,置于10 ml容量瓶中,用2 ml注射用水溶解,再加入流动相至刻度,摇匀,作为供试品溶液;②酸破坏:取LC约0.1 g,置于10 ml容量瓶中,用0.5 mol/L盐酸溶液2 ml溶解,1 h后用1 mol/L氢氧化钠中和,再加入流动相至刻度,摇匀,作为供试品溶液;③碱破坏:取LC约0.1 g,置于10 ml容量瓶中,用0.5 mol/L氢氧化钠溶液2 ml溶解,1 h后用1 mol/L盐酸中和,再加入流动相至刻度,摇匀,作为供试品溶液;④氧破坏:另取LC约0.1 g,置于10 ml容量瓶中,用过氧化氢溶液(3滴加水至25 ml)2 ml溶解,1 h后用加入流动相至刻度,摇匀,作为供试品溶液;⑤高温破坏:取LC原料约0.5 g,用注射用水10 ml溶解,于100℃水浴2 h,取2 ml置于10 ml容量瓶中,加入流动相至刻度,振摇混匀,作为供试品溶液;⑥光破坏:取LC原料约0.5 g,用注射用水10 ml溶解,强光照射4 h,取2 ml置于10 ml容量瓶中,加入流动相至刻度,振摇混匀,作为供试品溶液。
分别用选定的色谱条件进行试验,结果表明在该色谱条件下LC降解产生的各杂质峰与主成分峰能很好分离,分离度均>1.5,各破坏性试验样品色谱图见图1。
2.3.2 干扰试验 按处方取辅料,依供试品溶液制备法制备空白辅料溶液,分别取空白辅料溶液、空白流动相、LC对照品溶液、参比溶液c 20 μl,按2.1项下的色谱条件进行试验,结果见图2,辅料及流动相在LC对照品和杂质A对照品的保留时间处无干扰;LC与最近的杂质峰(杂质A)的分离度为1.895,LC的理论塔板数为1369,杂质A的理论板数为2417。
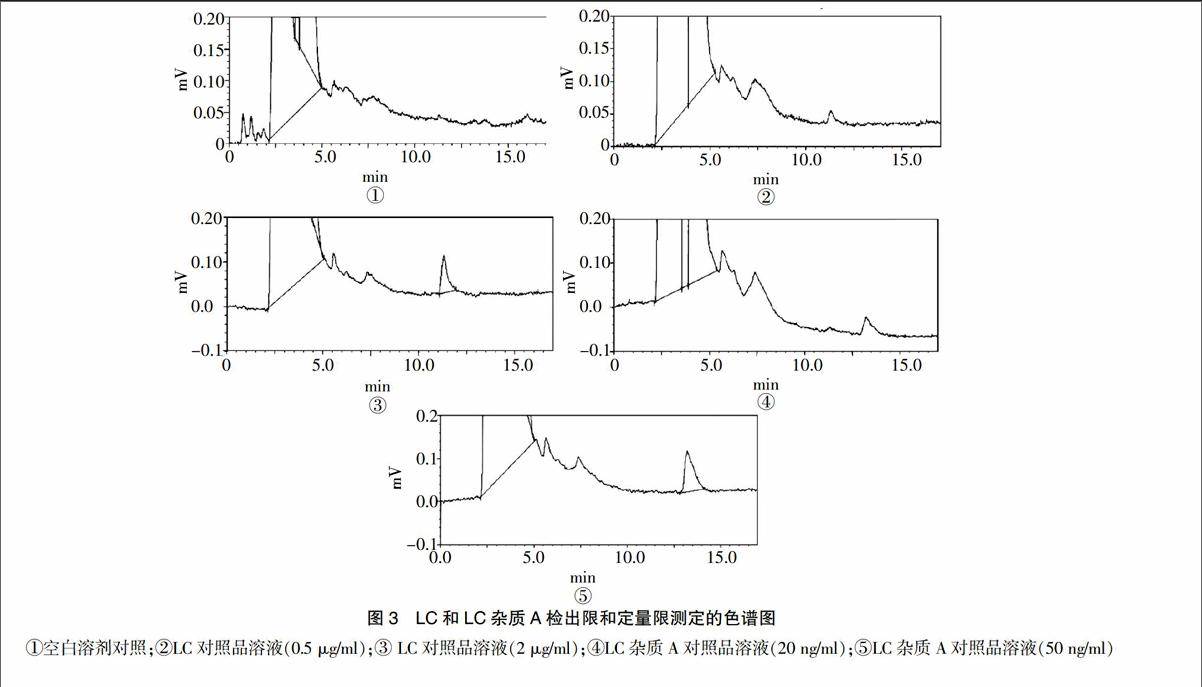
2.4 LC和LC杂质A的检出限和定量限
精密量取LC对照品溶液,逐步稀释配制系列浓度的溶液,分别进样测定,以信噪比(S/N)≥3 计算检出限,S/N≥10计算定量限;另将参比溶液a用流动相稀释成不同浓度溶液,同法测定检出限和定量限。结果LC的检出限和定量限分别为0.5、2.0 μg/ml,LC杂质A的检出限和定量限分别为20、50 ng/ml,色谱图见图3。
2.5 线性关系考察
2.5.1 LC线性关系考察 精密称取LC对照品适量,加流动相溶解并稀释至10 mg/ml,精密吸取上述溶液1.0、2.0、3.0、4.0、5.0、6.0、8.0 ml 置10 ml量瓶中,加流动相稀释至刻度,摇匀,分别吸取20 μl 注入液相色谱仪,记录色谱图。以进样浓度为横坐标,峰面积为纵坐标,将峰面积(y)对应LC浓度(x)进行回归,得方程:y=876.635 1091x+39 115.3624,r2=0.999 96(n=7)。LC进样浓度在1034~8274 μg/ml 范围内与峰面积呈良好的线性关系。
2.5.2 杂质A线性关系考察 取LC杂质A对照品适量,加流动相溶解并稀释至25 μg/ml,精密吸取上述溶液0.1、1.0、2.0、5.0、10.0 ml置10 ml量瓶中,加流动相稀释至刻度,摇匀,分别吸取20 μl 注入液相色谱仪,记录色谱图。以进样浓度为横坐标,峰面积为纵坐标, 将峰面积(y)对应杂质A浓度(x)进行回归,得方程:y=70 089.711 57x-7196.889 747,r2=0.999 87(n=5)。杂质A进样浓度在0.25~25 μg/ml 范围内与峰面积呈良好的线性关系。
2.6 精密度试验
精密称取LC对照品适量,加流动相溶解并稀释至5 mg/ml,摇匀备用。吸取20 μl注入液相色谱仪,记录色谱图,连续进样6次,记录LC和杂质A对应峰的峰面积,分别计算平均值和相对标准偏差(RSD),结果LC对应峰和杂质A对应峰峰面积平均值的RSD分别为0.38%、1.62%(n=6)。
2.7 稳定性试验
取2.6项下制作的溶液,分别在0、2、4、6、8 h测定,结果5次测定值基本不变,LC对应峰和杂质A对应峰峰面积平均值的RSD分别为0.093%、1.01%(n=5),显示样品溶液在8 h内稳定。
2.8 重复性试验
精密量取同一样品共6 份,分别加流动相溶解并稀释至5.0 mg/ml,测定含量,结果LC对应峰和杂质A对应峰峰面积平均值的RSD分别为0. 32%、1.83%,说明重复性良好。
2.9 LC加样回收率试验
依法配制LC 4.0、5.0、6.0 mg/ml 低、中、高3 种浓度溶液各3 份,每份分别按每支的处方量加入辅料,混匀,滤过,依法测定,LC平均回收率为101.29(n=9),RSD为0.55%,结果见表1,表明辅料对测定无干扰,回收完全。
2.10 LC杂质A加样回收率试验
依法配制LC杂质A 12.5、25.0、37.5 μg/ml 低、中、高3 种浓度溶液各3 份,每份分别按每支的处方量加入辅料,混匀,滤过,依法测定,杂质A平均回收率为100.35(n=9),RSD 为1.79 %,结果见表2,表明辅料对测定无干扰,回收完全。
2.11 样品含量测定
精密吸取LC注射液三个批次样品各1 ml,加流动相制成每毫升含5 mg 的溶液作为供试品溶液,取20 μl注入液相色谱仪,记录色谱图;另取2.2项下的LC对照品溶液和参比溶液a,同法测定,按外标法计算LC和杂质A的量,结果见表3。
3 讨论
在主药强酸、强碱破坏试验及强光光照试验以及高温试验过程中发现,在本实验HPLC色谱条件下,在相对于主峰保留时间1.20处能检出有关物质,经与LC杂质A对照品对比可知本杂质即为杂质A。为保证产品质量,欧洲药典[23]、美国药典[24]均对LC杂质A的限度做出规定,现有LC注射液国家药品标准[22]只对总杂质限度做出规定(自身对照法,<2%),未对已知杂质A的限度做出明确规定。文献[20]和本文2.3.2项数据均显示LC杂质A的响应值远大于LC,采用自身对照法单纯控制总杂质限度难以有效控制LC制剂的质量。此外,国家药品标准[16,22]规定的LC有关物质测定方法不能将LC和杂质A有效分开,我们曾尝试用C18柱来分析LC及杂质A,但两者分离度很小,改变流动相配比也难以将两者的分离度提高到0.9 以上而放弃。
根据LC在水溶液中显弱酸或弱碱性,在水溶液中分子带有一定电荷的特点,参考EP 6.0[23]收载的LC标准,我们选择强阴离子交换柱PhenoSphere 5u SAX 80A为分析介质,在流动相为乙腈∶磷酸盐(磷酸二氢钾6.81 g/L,以氢氧化钠溶液调节pH为4.7)=65∶35、流速1.0 ml/min的色谱条件下,LC和降解产生的各杂质峰得到有效分离,LC和最近杂质峰(杂质A)的分离度>1.5,辅料及流动相不干扰LC及杂质A的测定。
在该色谱条件下,LC的检出限和定量限分别为0.5、2.0 μg/ml,LC杂质A的检出限、定量限分别为20、50 ng/ml;LC在1034~8274 μg/ml 范围内线性关系良好,杂质A在0.25~25 μg/ml范围内线性关系良好 。
试验结果表明该方法简便、准确、专属,可用于LC注射液的含量测定和有关物质——杂质A的检测。
[参考文献]
[1] 朱宏明,刘青,房志仲.左卡尼汀的药理及临床应用研究进展[J].天津药学,2014,26(4):58-63.
[2] 苏庆丰,张林虎,张虹.左卡尼丁治疗心绞痛的临床疗效[J].中国现代医药杂志,2008,10(11):107-108.
[3] Moselhy SS,Demerdash SH.Serum free L-carnitine in association with myoglobin as a diagnostic marker of acute myocardial infarction[J].Clin Biochem,2009,42(1-2):78-82.
[4] 毛毳颖,郑雪冰,杨乐,等.左卡尼汀对急性心肌梗死心源性休克患者心功能及心肌酶的调节作用[J].中国实验诊断学,2009,13(9):1254-1256.
[5] 黎琳.左卡尼汀治疗慢性充血性心力衰竭的疗效观察[J].中国现代医药杂志,2011,13(7):56-58.
[6] 屈明玥.番茄红素拮抗氧化应激诱导的神经元损伤及分子机制研究[D].重庆:第三军医大学,2012.
[7] 唐敏.左卡尼汀联合重组人促红细胞生成素治疗肾性贫血的效果观察[J].中国当代医药,2014,21(14):109-110.
[8] 黄劲松,毛丹.左卡尼汀治疗肾性贫血30例疗效观察[J].吉林医学,2010,31(27):4721-4722.
[9] 付文静,邓英辉,张沛,等.左卡尼汀对维持性血液透析患者低血压的治疗作用[J].北京医学,2011,33(2): 131-133.
[10] Neri S,Pistone G,Saraceno B,et al.L-carnitine decreases severity and type of fatigue induced by interferon-alpha in the treatment of patients with hepatitis C[J].Neuropsychobiology,2003,47(2):94-97.
[11] 傅熙玲.左卡尼汀与肝病的相关性[J].中国医药科学,2011,1(6):30-31.
[12] Kajimoto Y,Kaneto H.Role of oxidative stress in pancreatic beta-cell dysfunction[J].Ann N Y Acad Sci,2004, 1011:168-176.
[13] 刘惠兰,张晓洁,曹峰.血液透析患者静脉应用左旋肉碱(可益能)的药代动力学研究[J].中国血液净化, 2003, 2(6):298-300,310.
[14] 梅长林,徐洪实,顾书华,等.血液透析病人血浆游离肉碱测定及临床意义[J].中华医学检验杂志,1998,21(6): 355-357.
[15] Fuller RK,Hoppel CL.Elevated plasma carnitine in hepatic cirrhosis[J].Hepathology,1983,3(4):554-558.
[16] 国家药典委员会.国家食品药品监督管理局·国家药品标准·新药转正标准·第64册[Z].2008:128-129.
[17] 曹玉,李萍,徐毅君,等.柱前衍生高效液相色谱法检测人血浆及尿液中左卡尼汀含量[J].中国医院药学杂志, 2008,28(15):1265-1268.
[18] 曹玉,李萍,徐毅君,等.糖尿病及其并发症患者血浆中左卡尼汀及其酰化物的含量分析研究[J].中国药理学通报,2014,30(7):952-955.
[19] 李庆宜,王学成,张超,等.柱前衍生化HPLC 法测定左卡尼汀中光学异构体的研究[J].中国药品标准,2011, 12(5):381-384.
[20] 高立军,王涛,全东琴.反相离子对高效液相色谱法测定左卡尼汀注射液中杂质A的含量[J].药物分析杂志,2013,33(7):1151-1156.
[21] 曾援.HPLC法测定左卡尼汀氯化钠注射液中左卡尼汀的含量[J].中国现代医药杂志,2010,12(6):63-65.
[22] 国家药典委员会.国家食品药品监督管理局·国家药品标准·新药转正标准[Z].第61册,2008:221.
[23] European Directorate for the Quality of Medicines & HealthCare.European Pharmacopoeia[Z].6th.2008:2257-2258.
[24] The United States Pharmacopoeia,USP32-NF27[Z].2009:2766-2768.
(收稿日期:2015-04-03 本文编辑:许俊琴)
