散发型嗜铬细胞瘤易感基因的热点区域的筛选
2015-08-07李汉忠
王 栋,李汉忠
(中国医学科学院 北京协和医学院 北京协和医院 泌尿外科, 北京 100730)
研究论文
散发型嗜铬细胞瘤易感基因的热点区域的筛选
王 栋,李汉忠*
(中国医学科学院 北京协和医学院 北京协和医院 泌尿外科, 北京 100730)
目的筛选与散发型嗜铬细胞瘤易感基因相关的染色体区域。 方法收集北京协和医院2011年9月至2013年5月临床诊断的42例散发型嗜铬细胞瘤病例。提取嗜铬细胞瘤及外周血的基因组DNA,用Sanger测序法检测遗传型嗜铬细胞瘤的突变基因;在证实为散发型的部分病例中,用单核苷酸多态性芯片(SNP 6.0 芯片)对其肿瘤组织及外周血的基因组进行扫描分析,精确定位可能的热点区域;然后以Q-PCR技术在其余的病例中进一步验证,从而最终确定散发型嗜铬细胞瘤相关的染色体片段。结果38例散发型病例,4例遗传型病例。通过SNP 6.0芯片扫描发现染色体片段的缺失较扩增更为常见,缺失的染色体包括 1p、3q、17p、22q和11p。1p上的chr1:74 957 006~86 132 879, chr1:58 096 424~67 700 471和chr1:98 902 091~107 622 4303个片段的缺失最为常见;Q-PCR验证后,上述3个片段仍为最常见的缺失区域。4例Ret突变者均出现上述3个片段的缺失。结论1号染色体短臂的部分区域上可能存散发型嗜铬细胞瘤相关的抑癌基因。
散发型嗜铬细胞瘤;单核苷酸多态性芯片;基因
嗜铬细胞瘤(pheochromocytoma, PCC)因分泌过量儿茶酚胺,可导致严重的心脑血管并发症,同时对于恶性PCC的诊断和治疗也缺乏理想的方法[1]。因而,明确PCC的发病机制,找到相关的突变基因以协助诊断及治疗就显得尤为迫切。目前发现NF1、RET、SDHx、TEME127以及MAX是遗传型PCC的易感基因[2]。但对于占多数比例的散发型PCC,虽有部分病例与上述基因相关,但仍有80%左右的病例其发病机制仍不明确[3]。基因芯片(gene chip)可在短时间内对全基因组进行扫描,具有快速、精确和低成本的分析检验能力,其中最新的单核苷酸多态性(sing1e nucleotidepolymorphism,SNP)芯片分辨率高,且能够同时检测DNA片断的扩增和缺失(copy-number variation, CNV)与等位基因的杂合性缺失(loss of heterozygosis, LOH),在相关基因筛选中的应用越来越广泛[4]。
1 材料与方法
1.1 病例和肿瘤标本
2011年9月至2013年5月共纳入临床诊断的散发型嗜铬细胞瘤42例,所有病例均无遗传性内分泌肿瘤综合征的家族史,且均未发现NF1、VHL综合征、多发性内分泌肿瘤病2型(MEN 2)及家族性嗜铬细胞瘤/副节瘤综合征伴随的其他肿瘤的证据。其中女性18人,男性24人,中位年龄43岁(17~61岁),单侧肿瘤39例,双侧肿瘤3例,肾上腺外9例,肾上腺区33例(表1)。
组织来源于手术切除的肿瘤标本和外周血。肿瘤标本离体后立即切割成直径1~2 mm的组织块,经液氮速冻后放入-80 ℃冰箱中保存。手术当日术前取头静脉血2.5 mL,放入肝素抗凝管内,置-80 ℃冰箱,作为肿瘤组织的正常配对。
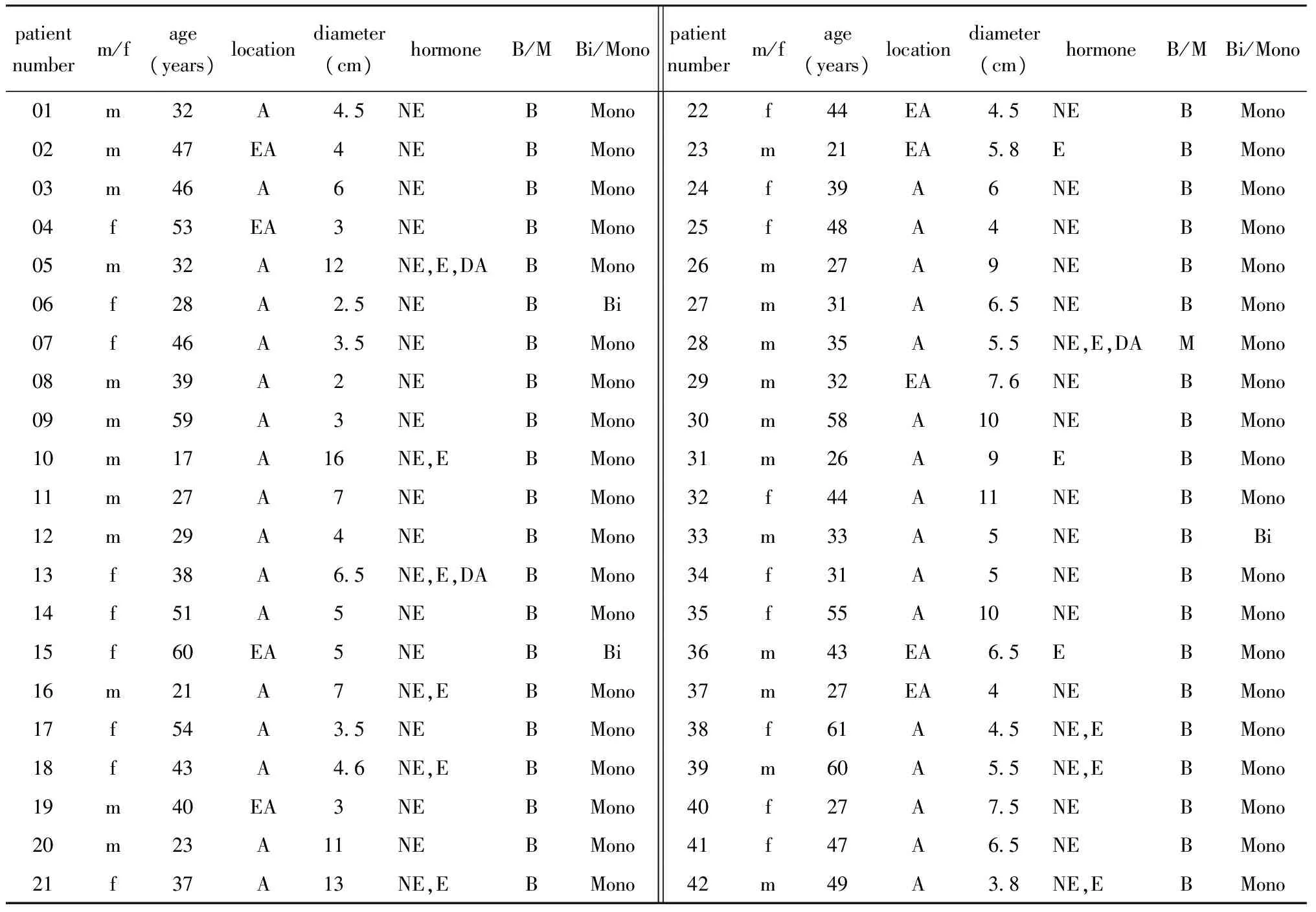
表1 临床资料
A.adrenal; EA.extra-adrenal; NE.norepinephrine; E.epinephrine; D.dopamine; B/M.benign/malignant; Bi/Mono.bilateral/monolateral.
1.2 基因组DNA的提取和质量控制
应用QIAamp DNA Blood Mini Kit提取全血基因组DNA,应用QIAamp DNA Mini Kit提取肿瘤组织基因组DNA,均按生产商的说明进行操作。应用于芯片实验的基因组DNA样本必须符合以下质控条件:用Nanodrop分光光度计检测吸光度,A260/A280应接近1.8,浓度大于500 μg/μL。用1%琼脂糖凝胶电泳出现1条明亮单1条带,无明显拖尾。
1.3 Sanger测序
PCR扩增血液及肿瘤组织基因组DNA中各个PCC相关基因(RET,VHL,SDHB,SDHC和SDHD)的外显子片段,应用Sanger测序检测是否存在这些基因的突变。MEM127和MAX突变在PCC患者中的比例低于1%,因而未检测这两种基因,而NF1因可通过特异临床的表现而排除,故也未检测NF1的突变。
1.4 SNP芯片检测及热点区域的初步定位
在证实为散发型的病例中,应用SNP6.0芯片(Affymetrix公司提供)扫描随机抽取的14例患者的基因组DNA(包括肿瘤组织和外周血的白细胞)。实验过程按照Affymetrix® Genome-Wide Human SNP Nsp/Sty 6.0 User Guide芯片实验操作手册进行。
前述程序结束后,以3000 7G扫描仪扫描芯片,将原始数据文件输入到Partek Genomics Suite 软件,肿瘤样本的拷贝数以配对的外周血为基线进行分析,拷贝数大于2.3和小于1.6分别作为扩增和缺失的标准,其中技术参数严格限制:基因组marker数>10;P<0.001;信噪比>0.3。选取CNV频率最高的染色体片段作为相关的热点区域进行进一步验证。
1.5 Q-PCR验证染色体易感区域
通过Q-PCR在其余的散发型病例中验证SNP6.0芯片所初步确定的热点区域。首先根据NCBI Genebank及相关文献,确定热点区域所对应的DNA序列,进而设计出引物。然后配制Q-PCR反应体系,进行PCR反应。Q-PCR反应在StepOne Plus实时荧光定量PCR仪上进行(表2,表3)。以全部血液基因组DNA的混合物为拷贝数正常对照样本。以C2基因片段作为标化序列,应用2-ΔΔCt法计算相对拷贝数。将扩增的PCR产物进行溶解曲线分析判断扩增产物的特异性。相对拷贝数小于1.4认定为发生缺失。为使Q-PCR产物能代表所检测的DNA片段,为每个片段分别设计3组引物。同一样本3组引物的结果一致可认定为发生缺失。

表2 配制的Q-PCR反应体系
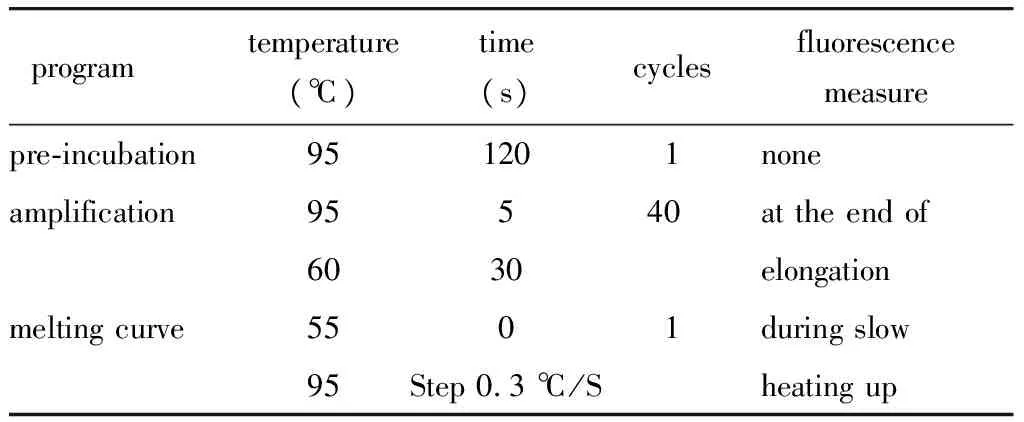
表3 Q-PCR扩增反应条件
使用StepOneTM软件(荧光定量操作及数据分析软件)对数据进行处理。将超过全部散发型病例数50%的区域确定为散发型PCC相关的染色体区域。临床数据与CNV片段之间的关联通过χ2检验来验证。
2 结果
2.1 基因组DNA提取结果
肿瘤及外周血白细胞DNA的凝胶电泳显示DNA条带清晰单一,吸光度A260/280比值均在1.7~1.9之间,证实提取的基因组DNA为高纯度、无降解(图1)。
2.2 Sanger测序的结果
42例病例中,4例外周血的突变检测为阳性,其中1例为VHL突变,3例为RET突变(图2)。其余38例外周血突变检测的结果均为阴性,证实为散发型PCC,其中1例(41号)肿瘤组织的RET基因显示为阳性。

图1 部分病例中肿瘤组织与外周血的基因组DNA凝胶电泳结果
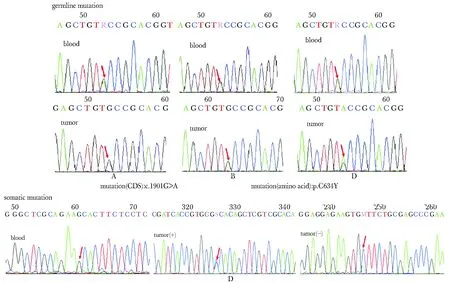
A,B,C. Sequencing consequences of DNA products from blood tissues indicated a heterozygous G/A mutation at codon 634, which was consistent with the results of DNA from tumor tissues sequencing; D. Sequencing consequences of DNA products from tumor tissues indicated a heterozygous C/A mutation at codon 618, which was not conformed in the results of DNA from blood tissues sequencing
图2 Sanger测序的结果(ret基因)
Fig 2 The result of Sanger sequence mutation in ret gene
2.3 染色体热点区域的初步确定
经SNP6.0芯片扫描,在14例散发型PCC中均发现了CNV。其中,缺失的染色体包括1p(12/14)、3q(8/14)、17p(4/14)、22q(4/14)和11p(4/14),扩增的包括22q(2/14)、17q(3/14)和9q(2/14)(表4,图3)。CNV频率最高的染色体为1p,其上的3段DNA片段chr1:74 957 006~86 132 879, chr1:58 096 424~67 700 471和chr1:98 902 091~107 622 430的缺失频率均超过50%(表4,图4)。另外4例遗传型病例的SNP芯片扫描结果显示, RET基因突变的3例均发生了1p中片段的缺失(表4)。
2.4 Q-PCR 结果
根据NCBI Genbank 及相关文献,为上述3各片段对应的DNA序列分别设计3组引物(表5)。熔解曲线显示Q-PCR产物均为单一扩增(图5)。经Q-PCR验证,chr1:74 957 006~86 132 879, chr1:58 096 424~67 700 471及 chr1:98 902 091~107 622 430这3个片段发生的频率分别为28/38、27/38及23/38,均超过50%(表4)。
3 讨论
目前通过比较基因组杂交技术(comparative genomic hybridization,CGH)发现发现散发型PCC中CNV很常见,拷贝数缺失是CNV最常见的表现形式[5]。拷贝数缺失常导致等位基因杂合性的缺失(LOH),而LOH的区域往往是抑癌基因所在的位置[6]。这些研究结果高度提示在缺失的染色体片段中可能存在着散发型PCC的抑癌基因。但CGH只能得出CNV的位置,而无法确定是否存在LOH,因而无法确定相关基因的位置。而SNP芯片可以在确定CNV区域的同时验证是否存在LOH[4]。本研究通过SNP6.0芯片共扫描的14例散发型PCC均发生了CNV。其中扩增的7例,而全部14例均出现了缺失,这表明拷贝数缺失是散发型PCC发病过程的重要事件,也提示抑癌基因的失活可能在散发型PCC的发病中发挥了重要的作用[7]。另外本研究发现缺失的染色体包括1p,3q,11p,13q,17p和22q,这与既往的研究结果类似[7- 8]。
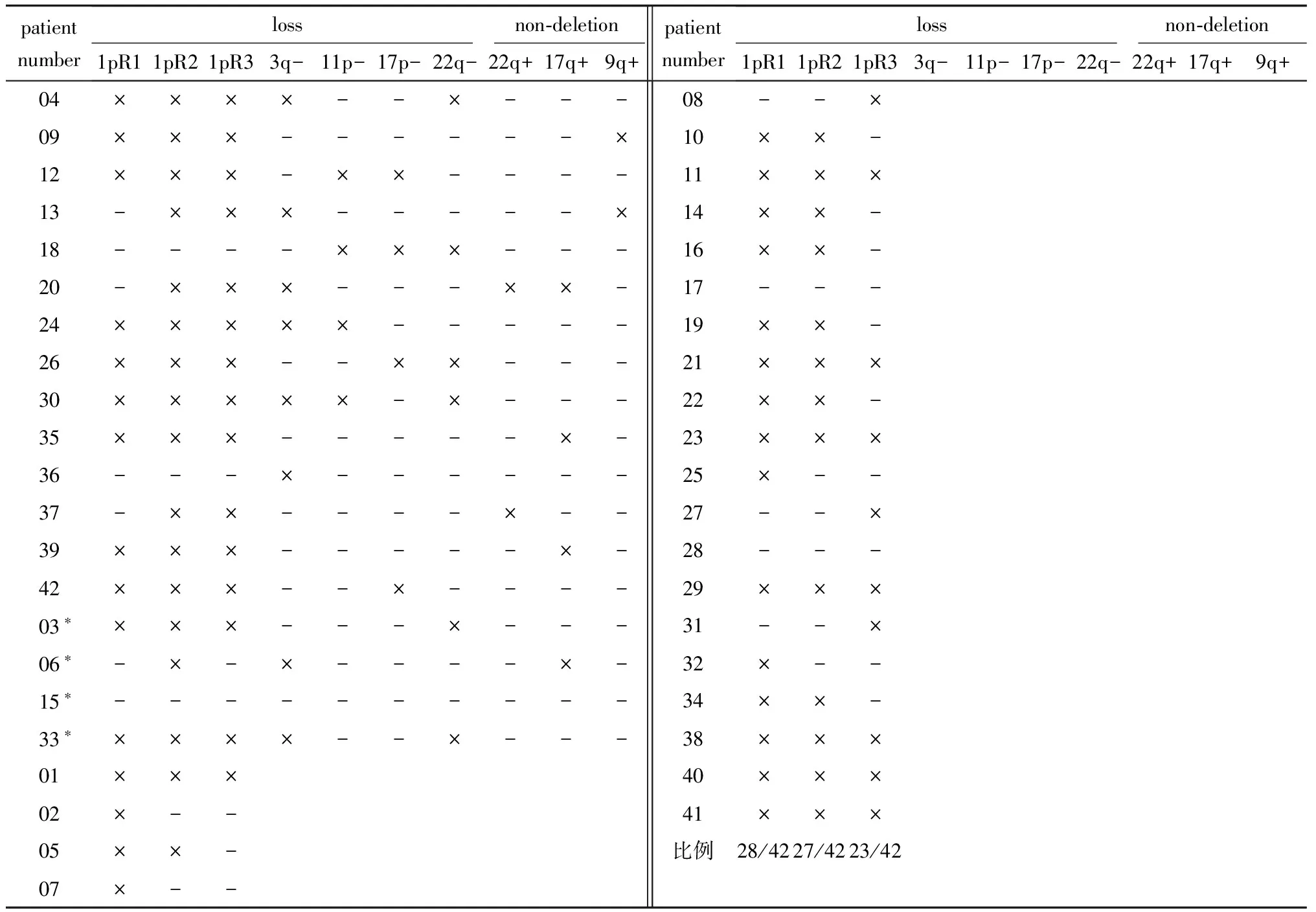
表4 SNP芯片及Q-PCR结果
grey area.result of SNP chip;*inherited cases;×.finding the deletion;-.no foundong the deletion;blank.no data.

表5 以3段热点区域设计的Q-PCR引物
region1.chr1.74 957 006-86 132 879;region2.chr1:58 096 424-67 700 471;region3.chr1:98 902 091-107 622 430.
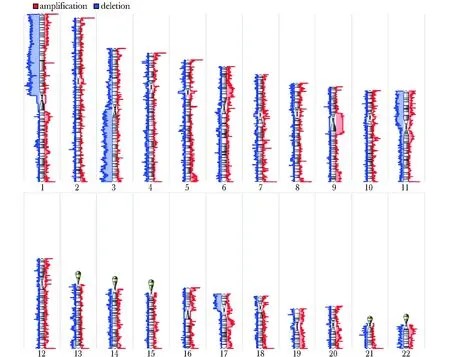
Green lines on left side indicate loss of DNA copies situated in that area; Red lines on right side indicate gains of DNA copies situated in that area
图3 散发型PCC中CNV的频率
Fig 3 The frequency of CNV in sporadic PCC
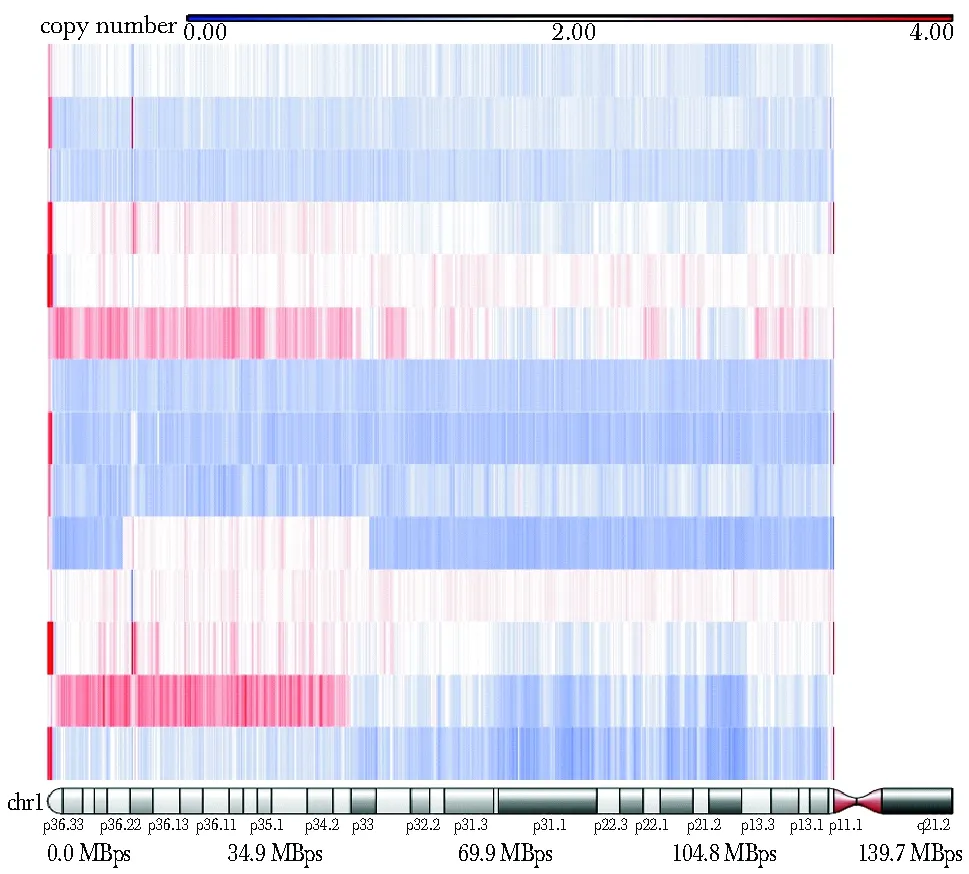
Blue regions indicate deletion area图4 SNP6.0芯片显示的14例散发型PCC中1p上CNV的频率Fig 4 The frequency of CNV of 1p in sporadic PCC
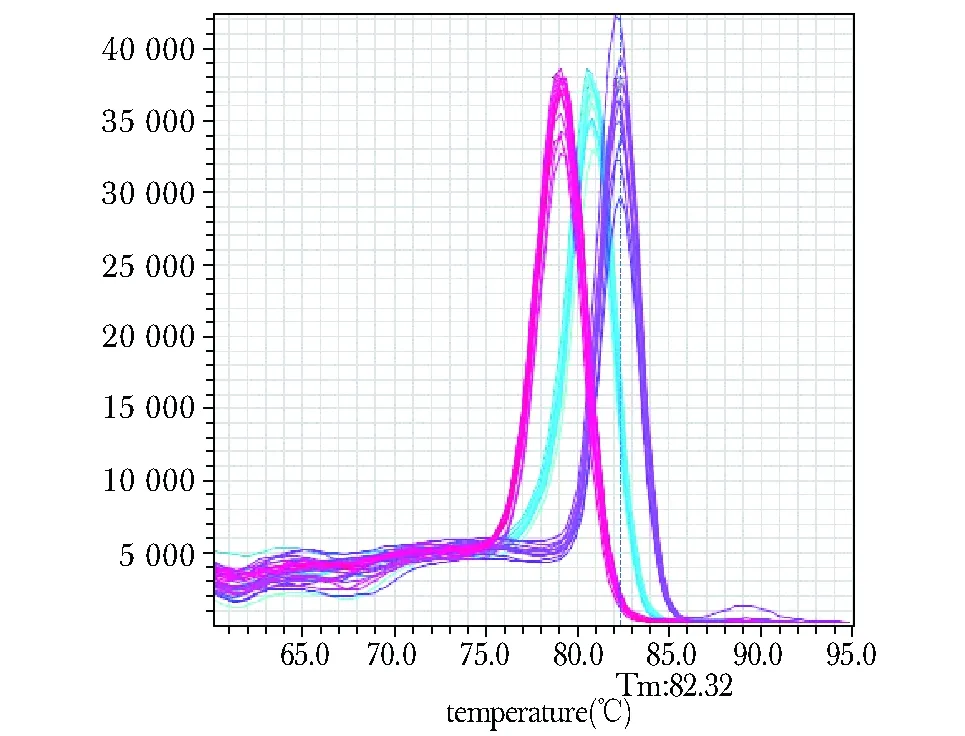
The single peak indicates single product图5 Q-PCR产物的熔解曲线Fig 5 Melt curve of Q-PCR
目前认为1p是散发型PCC最常见的缺失片段[5],1p的缺失与多种肿瘤相关,如少突胶质细胞肿瘤、神经母细胞瘤[9- 10]。本研究中最常见的缺失片段也是1p(12/14),并且发现了其上3段最常见的缺失区域,结合Q-PCR的验证结果,提示这3段区域存在散发型PCC的相关基因。
另有研究发现RET基因突变的遗传型PCC中,1p的缺失比率高达80%~100%,而MEN2中PCC的外显率却仅有5%[11],这提示除RET基因突变外,尚需其他DNA的异常事件才能最终导致PCC。本研究中4例RET突变的病例(3例遗传型,1例散发型)均发生了1p片段的缺失,这提示RET突变的散发型和遗传型PCC具有类似的发病机制,除了已知的RET基因突变外,还可能与1p部分区域的缺失相关。
本研究为初步实验结果,通过深入研究散发型与遗传型PCC以及良恶性PCC之间有表达差异的基因, 可以进一步探索PCC的分子病因学基础,从而为PCC的早期诊断、良恶性鉴别以及治疗提供新的途径。
[1] Tsirlin A, Oo Y, Sharma R,etal. Pheochromocytoma: a review[J].Maturitas. 2014, 77:229- 238.
[2] Galan SR, Kann PH. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma[J].Clin Endocrinol, 2013, 78:165- 175.
[3] Lenders JW, Eisenhofer G, Mannelli M,etal[J].Phaeochromocytoma[J]. Lancet, 2005, 366:665- 675.
[4] Gruel N, Benhamo V, Bhalshankar J,etal. Polarity gene alterations in pure invasive micropapillary carcinomas of the breast[J].Breast Cancer Res,2014,16:R46. [Epub ahead of print]
[5] Dannenberg H, Speel EJ, Zhao J,etal. Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas [J].Am J Pathol, 2000, 157:353- 359.
[6] Couto SS. The pathologist’s slide reveals more than meets the eye: loss of heterozygosity and cancer biology [J].Vet Pathol, 2011, 48:236- 244.
[7] Sandgren J, Diaz de Ståhl T, Andersson R,etal. Recurrent genomic alterations in benign and malignant pheochromocytomas and paragangliomas revealed by whole-genome array comparative genomic hybridization analysis[J].Endocr Relat Cancer, 2010, 17:561- 579.
[8] Sun HY, Cui B, Su DW,etal. LOH on chromosome 11q, but not SDHD and Men1 mutations was frequently detectable in Chinese patients with pheochromocytoma and paraganglioma [J].Endocrine, 2006, 30:307- 312.
[9] 熊佶,刘 颖,李 超.少突胶质细胞瘤染色体 1p/19q 杂合性缺失与 P53 蛋白表达的相关性研究[J]. 中华病理学杂志, 2009:123- 125.
[10] Grundy PE, Breslow NE, Li S, Perlman E,etal. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms[J].J Clin Oncol, 2005, 23:7312- 7321.
[11] Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes[J].Horm Metab Res, 2012,44:328- 333.
Screen of the tumor-related regions of sporadic pheochromacytoma
WANG Dong, LI Han-zhong*
(Dept. of Urology, PUMC Hospital, CAMS & PUMC, Beijing 100730, China)
Objective To identify candidate regions of sporadic pheochromacytoma(PCC). Methods Totally 42 patients who were clinically diagnosed as sporadic PCC from 2011-9 to 2013-5 in PUMCH were enrolled. To extract whole genome DNA from their tumors as well as peripheral blood leukocytes and to exclude inherited cases by Sanger sequence mutation. Within 14 verified cases of sporadic PCC, we applyed single nucleotide polymorphism (SNP) chip to detect whole genome DNA copy number variations (CNV) and loss of heterozygosity (LOH) to initially locate the hot regions. Finally apply Q-PCR to confirm the hot regions in left cases. Results 38 cases were identified as sporadic PCC, and 4 as inherited cases. CNV were found in 14/14 tumors, of which deletions were more common. Missing regions occurred in 1p,3q,17p,22q,11p. On the other hand, of the 3 inherited cases, deletion was also detected. The loss of parts of arm 1p is the most common, including chr1:74 957 006~86 132 879, chr1:58 096 424~67 700 471, and chr1:98 902 091~107 622 430. The result of Q-PCR confirmed the above-mentioned three regions, and the three segments are final candidate regions. Conclusions Partially deletion of 1p in most cases is the most striking phenomenon, we believe that the deletion of 1p may occur with PCC development, and there may be some tumor suppressor gene(s) within these areas.
sporadic pheochromacytomas; single nucleotide polymorphism chip; genes
2014- 07- 03
2014- 10- 08
1001-6325(2015)01-079-07
R699.3
A
*通信作者(corresponding author):jellowaaa7@163.com
