狂犬病病毒单质粒拯救系统的建立及应用
2015-07-21孙玉章许运斌丛彦龙KarlKlausConzelmann
孙玉章,何 彪,许运斌,丛彦龙,Karl-Klaus Conzelmann
(1.中国动物卫生与流行病学中心,青岛 266032;2.军事医学科学院军事兽医研究所,长春 130122;3.浙江大学动物科学学院,杭州 310020;4.吉林大学动物医学学院,长春 130062;5.慕尼黑大学基因中心,慕尼黑81377)
狂犬病(rabies)是由狂犬病病毒(rabies virus,RABV)引起的以侵犯中枢神经系统为主的急性人兽共患传染病,病死率几乎高达100%。据世界卫生组织(World Health Organization,WHO)报道全球每年有约55000人死于狂犬病,其中绝大多数发生在亚洲、非洲等发展中国家[1]。狂犬病病毒是弹状病毒科(Rhabdoviridae)狂犬病病毒属(Lyssavirus)成员,其基因组为不分节段的负链RNA病毒。RABV基因组编码5种结构蛋白,依次为核蛋白(nucleoprotein,N)、磷酸化蛋白 (phosphoprotein,P)、基质蛋白(matrix protein,M)、糖蛋白(glycoprotein,G)和RNA依赖的RNA聚合酶(RNA dependent RNA polymerase,RdRp/L)[2]。目前,学术界一般将狂犬病病毒属划分为7个基因型,其中6个基因型的RABV均以蝙蝠为宿主和媒介生物(基因III型,即MOKV的确切宿主尚未确定),另外还有一些新近分离出的RABV毒株尚待分型[3]。
狂犬病病毒基因组RNA只有与核蛋白形成核糖核蛋白复合物(ribonucleoprotein,RNP)才能在磷蛋白的催化下被RNA依赖的RNA聚合酶蛋白识别并作为转录和复制的模板[4]。因此,经典的RABV的反向遗传操作系统除了构建具有精确末端序列的全长基因组cDNA克隆外,还需要构建表达核蛋白、磷蛋白和聚合酶大蛋白的真核表达质粒共同转染适当的细胞系才可能获得具有感染性的病毒颗粒[5]。
本研究在已建立SAD L16株高效拯救的反向遗传操作平台基础上[6],以SAD L16株全长基因组cDNA克隆为骨架,利用内部核糖体进入位点(internal ribosome entry site,IRES)分别构建了5个全长cDNA克隆[7]并直接转染BSR T7/5细胞系,获得了2株重组病毒。通过进一步鉴定和分析重组狂犬病病毒株的生物学特性,本研究首次建立起RABV的单质粒拯救系统,并以此为平台开展了一系列研究。
1 材料与方法
1.1 病毒与细胞
BSR T7/5细胞系由本室保存,以含10%胎牛血清的GMEM培养。狂犬病病毒SAD L16株由本室保存,其全长cDNA克隆质粒pSAD-L16由本室构建并保存。分别克隆有脑心肌炎病毒(encephalomyocarditis virus,EMCV)IRES 序列、脊髓灰质炎病毒(polivirus,PV)IRES序列和明脉扁刺蛾病毒(Thosea asigna virus,TaV)2A序列的pRLCMV载体质粒均由本室构建并保存。
1.2 主要试剂
Phusion DNA聚合酶购自Finnzymes公司,反转录聚合酶购自Roche公司,T4DNA连接酶购自NEB公司,质粒快速抽提试剂盒和凝胶快速回收试剂盒均购自QIAGEN公司,转染试剂Lipofectamine 2000购自Invitrogen公司,CaPO4转染试剂盒购自Stratagene公司,其他限制性内切酶主要购自NEB或Fermentas公司,细胞培养血清及相关试剂均购自Invitrogen公司,其他试剂或化学药品均购自Invitrogen、Roche或Sigma公司。
1.3 重组全长cDNA克隆的构建
根据全长cDNA感染性克隆pSAD-L16 N、P和L基因上游序列的不同分别设计多对引物扩增EMCVIRES 序列(e)、PVIRES 序列(p)和 TaV 2A序列(t),将其分别克隆入pSAD-L16载体的对应位置中,构建成5个全长cDNA克隆:pSAD-eNePeL(Ⅰ)、pSAD-eNtPeL(Ⅱ)、pSAD-NeNePeL(Ⅲ )、pSAD-NeNpPeL (Ⅳ )和 pSAD-NeNtPeL(Ⅴ)。所有构建的cDNA克隆经测序正确后大量制备并纯化。5个全长cDNA克隆的构建策略如图1所示。
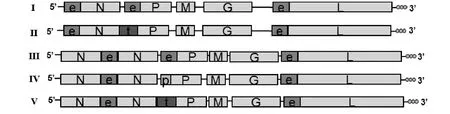
图1 重组全长cDNA克隆构建模式Fig.1 Construction of 5full-length cDNA clones based on pSAD-L16
1.4 重组病毒的拯救
转染前16h将BSR T7/5细胞分铺到6孔板,用含10%胎牛血清的GMEM培养。转染前1h更换为不含血清的GMEM培养。CaPO4法分别转染10μg全长cDNA克隆质粒至每孔细胞中,轻晃混匀。同时设单质粒组(分别单独辅助转染pTiT-N、pTiT-P或pTiT-L质粒)和常规组(同时转染pTiTN、pTiT-P和pTiT-L 3个辅助质粒)4个对照组。37℃培养6h,PBS清洗3次后更换含10%胎牛血清的GMEM继续培养。转染后每3d取培养液感染新的BSR T7/5细胞并以FITC标记的鼠抗狂犬病病毒N蛋白单克隆抗体进行检测,直到第12天。获救的每一代次的重组狂犬病病毒首先进行RTPCR并测序,序列完全正确的病毒测定滴度后以MOI=0.01接种于T75细胞培养瓶,每72h收获一次培养液,连续收获3次,并储存于-80℃超低温冰箱。
1.5 重组狂犬病病毒生物学特性的鉴定
重组狂犬病病毒的滴度测定:将收获的细胞培养上清液以不含胎牛血清的GMEM连续做10倍梯度稀释后取100μL接种于96孔板中的单层BSR T7/5细胞,于37℃培养48h,弃细胞上清液,PBS清洗后用80%丙酮冷藏固定20min,洗弃冷丙酮后以FITC标记的鼠抗狂犬病病毒N蛋白单克隆抗体37℃染色2h,PBS清洗3次后置于荧光显微镜下观察并计数,计算病毒终释滴度。
生长动力学曲线的测定:拯救重组病毒以MOI=0.01接种于T25细胞培养瓶,6h后收集细胞培养液并更换新鲜GMEM培养液,每24h收获一次,直到96h。收获的细胞培养液梯度稀释后接种于96孔板,37℃培养48h后以FITC标记的鼠抗狂犬病病毒N蛋白单克隆抗体进行检测,计算终释病毒滴度并绘制多步生长曲线。
重组狂犬病病毒的遗传稳定性:拯救重组病毒以MOI=1接种于BSR T7/5细胞,每96~120h收获细胞培养上清液,分别进行转移到新BSR T7/5细胞中继续传代和转染96孔板测定病毒滴度,连续传代20次,每5代收集一次细胞培养上清液进行RT-PCR并测序。
2 结 果
2.1 重组狂犬病病毒的拯救
在完全不依靠转染辅助质粒的情况下,只有pSAD-NeNePeL(Ⅲ)和pSAD-NeNtPeL(Ⅴ)能够成功拯救出重组狂犬病病毒,pSAD-NeNePeL直接转染成功率较高(12/12),pSAD-NeNtPeL 成功率较低(8/14),单独转染任何一种辅助质粒pTiT-N、pTiT-P或pTiT-L对拯救效率均无显著影响,转染后第6天均可在细胞培养上清液中检测到病毒颗粒(图2)。
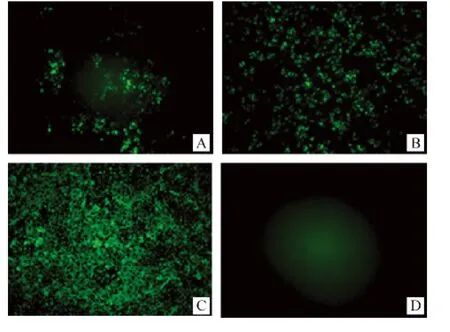
图2 重组狂犬病病毒的DFA检测Fig.2 Identification of recombinant RABVs with FITC-antiN-Mab
2.2 重组狂犬病病毒的增殖动态
初代获救的重组狂犬病病毒的多步生长曲线如图3所示,重组狂犬病病毒SAD-NeNePeL和SADNeNtPeL均高度致弱且增殖缓慢。

图3 重组狂犬病病毒多步生长曲线Fig.3 Multistep growth curves of recombinant RABVs
SAD-NeNePeL和 SAD-NeNtPeL 2株重组狂犬病病毒在终释滴度均能形成与野生型亲本毒株SAD-L16株大小形态不同的荧光灶(图4)。
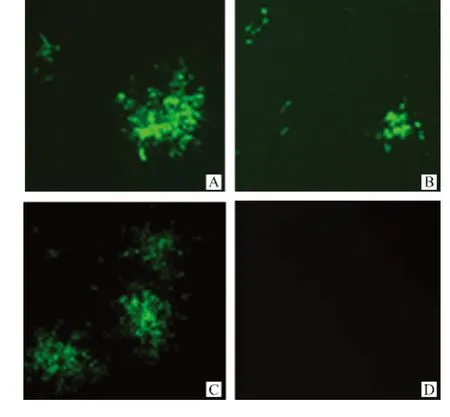
图4 重组狂犬病病毒的荧光灶形态Fig.4 Different phenotypes in foci size of recombinant RABVs
2.3 重组狂犬病病毒的遗传稳定性
重组狂犬病病毒SAD-NeNePeL在BSR T7/5细胞中仅能传3~5代,RT-PCR结果显示在第3代的细胞培养液上清中就已经存在不同水平的重组株,且表现出不同的生长动力学特性(未发表数据)。重组狂犬病病毒SAD-NeNtPeL在BSR T7/5细胞中则能稳定传代20次,且RT-PCR结果显示序列高度稳定。
3 讨 论
经典狂犬病病毒反向遗传系统的建立除需精确构建病毒基因组的全长cDNA克隆外,还需提供分别表达N、P和L 3种蛋白质的辅助质粒,才可能形成RNP复合物并起始病毒基因组的转录和翻译。顺式作用元件IRES序列的发现[8-9]给我们的研究提供了另外一种可能:通过在狂犬病病毒基因组全长cDNA克隆中N、P和L基因上游克隆入相应序列以实现单一质粒同时表达多种mRNA。因此,我们分别设计了包含EMCVIRES序列、PVIRES序列或TaV 2A序列的多个全长cDNA克隆,并先后建立起了三质粒拯救系统和二质粒拯救系统,最终有2个单质粒拯救系统成功拯救出重组狂犬病病毒株。
单质粒拯救系统在负链分节段的流感病毒已有报道[10],但其采用的方法并不适用于负链不分节段的狂犬病病毒。通过引入顺式作用元件IRES序列建立的单质粒拯救系统获救的SAD-NeNePeL株具有较高的拯救成功率,但基因组间的重配概率较高,进一步的研究发现在细胞培养上清液中至少存在3种基因重组株且呈现不同的生长动力学特性;而SAD-NeNtPeL则可以稳定的表达多顺反子mRNA且具有相当的遗传稳定性。值得注意的是在N基因上游引入IRES序列构建的多个全长cDNA克隆无一拯救成功,进一步的研究发现导致拯救失败的原因是在N基因氨基端存在一个此前未曾报道过的顺式调控元件(数据未发表),因此本研究通过引入表达更高效的EMCVIRES序列串联起2个N基因,最终无需提供辅助质粒pTiT-N即可拯救出重组狂犬病病毒。
利用建立的狂犬病病毒单质粒拯救系统,本室先后开展了基于Pol-II聚合酶拯救系统的建立以及更高效的二质粒拯救系统的建立等研究。这些反向遗传操作系统的建立为深入研究狂犬病病毒基因组结构与功能、狂犬病病毒分子致病机制、狂犬病病毒与宿主相互作用以及新型嗜神经性病毒载体和转基因动物模型等提供了一个有力的研究工具和基础的技术平台。
(References):
[1]KNOBEL D L,CLEAVELAND S,COLEMAN P G,et al.Re-evaluating the burden of rabies in Africa and Asia[J].Bull World Health Organ,2005,83(5):360-368.
[2]TAKAYAMA-ITO M,INOUE K,SHOJI Y,et al.A highly attenuated rabies virus HEP-Flury strain reverts to virulent by single amino acid substitution to arginine at position 333in glycoprotein[J].Virus Res,2006,119(2):208-215.
[3]DIETZGEN R D,KUZMIN I V.Rhabdoviruses:molecular taxonomy,evolution,genomics,ecology,hostvector interactions,cytopathology and control[M].London:Caister Academic Press,2012:37-57.
[4]SHOJI Y,INOUE S,NAKAMICHI K,et al.Generation and characterization of P gene-deficient rabies virus[J].Virology,2004,318(1):295-305.
[5]SCHNELL M J,MEBATSION T,CONZELMANN K K.Infectious rabies viruses from cloned cDNA[J].EMBO J,1994,13(18):4195-4203.
[6]GHANEM A,KERN A,CONZELMANN K K.Significantly improved rescue of rabies virus from cDNA plasmids[J].Eur J Cell Biol,2012,91(1):10-16.
[7]MARSCHALEK A,FINKE S,SCHWEMMLE M,et al.Attenuation of rabies virus replication and virulence by picornavirus internal ribosome entry site elements[J].J Virol,2009,83(4):1911-1919.
[8]PIJLMAN G P,ROODE E C,FAN X,et al.Stabilized baculovirus vector expressing a heterologous gene and GP64from a single bicistronic transcript[J].J Bio-technol,2006,123(1):13-21.
[9]WAGSTAFF M J,LILLEY C E,SMITH J,et al.Gene transfer using a disabled herpes virus vector containing the EMCV IRES allows multiple gene expression in vitro and in vivo[J].Gene Ther,1998,5(11):1566-1570.
[10]ZHANG X,KONG W,ASHRAF S,et al.A one-plasmid system to generate influenza virus in cultured chicken cells for potential use in influenza vaccine[J].J Virol,2009,83(18):9296-9303.
