表观遗传学及现代表观遗传生物医药技术的发展
2015-07-19姜楠潘学峰
姜楠潘学峰,2
(1. 北京理工大学生命学院,北京 100081;2. 河北大学基础医学院,保定 071002)
表观遗传学及现代表观遗传生物医药技术的发展
姜楠1潘学峰1,2
(1. 北京理工大学生命学院,北京 100081;2. 河北大学基础医学院,保定 071002)
表观遗传学和基于表观遗传机制的生物医药技术的研究已经成为后基因组时代生命科学技术领域的重要组成部分。围绕肿瘤、心脑血管疾病、糖尿病及中老年神经退行性疾病等过程中DNA甲基化修饰、组蛋白翻译后修饰及非编码RNA等表观遗传学改变的深入研究,不仅有利于理解相关疾病的分子病理机制,而且,更有助于探寻基于表观遗传机制的有效治疗手段。在阐释表观遗传学修饰机制的基础上,对疾病过程中异常的表观遗传学修饰及相关生物医药技术的研究现状进行了归纳总结。
表观遗传学;DNA甲基化;组蛋白翻译后修饰;非编码RNA;人类疾病
1 表观遗传学的缘起
1942年,Conrad H. Waddington 依据胚胎学的“epigenesist”(渐成说,该学说强调生物个体发育和分化是基于细胞内的可溶性组分所能发生的化学反应)和“Genetics”(遗传学)创建了“Epigenetics”一词,用于描述胚胎发育过程中基因及其产物的“临时性”作用所能呈现出的生物性状(Phenotype),以及基于这些性状而最终发育成成熟生物个体的一个生物学分支。最初,“Epigenetics”一词汉译为“实验胚胎学”,故“Epigenetics”的本意是指受精卵在不改变核内基因组的前提下,通过基因和基因编码产物之间的相互作用执行“发育”和“分化”,并最终出现高度复杂且成熟的生物个体的机制。随后,“Epigenetics”的定义几经改变,但近年来的研究进展再次表明,“表观遗传学”的研究内容又重新回归了当初其基于早期发育和分化的定义。
但是,如今的“表观遗传学”已经承载了更多的分子生物学语义,已经成为研究生物个体在保持细胞内DNA完整的情况下,通过包括DNA的“化学修饰”(chemical modification of DNA)、组蛋白翻译后修饰及非编码RNA等途径,通过改变染色质结构来塑造“环境-基因互作”所要求的基因表达模式的一个崭新的现代分子生物学分支学科。
事实上,多细胞生物个体内的“体细胞”均含有相同的DNA组分,但实现“分化”和“发育”成为不同类型的体细胞内的“基因表达模式”(patterns of gene expression)却体现明确的差异。由这些差异“表达模式”所确定的性状可以稳定地表现出“克隆性遗传”(clonally inherited)现象。据此有人为“表观遗传学”赋予了一个比较模糊的定义,即研究有丝分裂和/或减数分裂的可遗传的基因功能的改变,这些基因功能的改变不能通过改变DNA序列加以解释的遗传学。
本文将围绕癌症、心脑血管疾病、糖尿病及神经退行性疾病等重大疾病过程中DNA的化学修饰、组蛋白翻译后修饰及非编码RNA等表观遗传学修饰机制等研究热点及相应的现代医药生物技术研发现状展开分析和讨论。
2 表观遗传修饰的主要手段
在动植物等多细胞生物个体中,一个细胞所拥有的特定“身份”取决于该细胞内DNA调控元件与蛋白质调控复合体之间相互作用所建立的独特的表观遗传学修饰状态。本质上,这些表观遗传修饰状态对应着由染色质中的DNA、组蛋白等的修饰,以及由染色质重塑所建立的独特的基因表达模式类型。不仅如此,表观遗传学修饰还直接影响着该细胞内包括DNA复制、DNA损伤修复、重组、染色体压缩和分离等几乎所有与DNA代谢有关的过程。自20世纪90年代以来,表观遗传学研究取得了长足进展。现已明确了DNA特定位点处的甲基化修饰、核小体中组蛋白翻译后修饰、非编码RNA调控、组蛋白变体及受催化的染色质重塑等各种表观遗传修饰类型[1]。
2.1 DNA甲基化与去甲基修饰
DNA甲基化修饰是发生在DNA上的一类持久且相对稳定的表观遗传修饰,它的主要生物学功能是在染色质水平通过影响转录因子与DNA的接触对基因转录加以调节[1]。几十年的研究表明,DNA甲基化修饰在遗传印记(genetic imprinting)[2]、X-染色体失活、抑制基因组内重复DNA序列的不稳定性和转座子转录方面发挥着关键的作用[3]。
真核生物DNA分子中某些位点处的胞嘧啶的5'位置是常见的甲基化修饰(5-Methylcytosine,5-meC)部位。出现在相应部位的胞嘧啶甲基化修饰是表观遗传修饰促使基因沉默(epigenetic gene silencing)和染色质成为“异染色质”(heterochromatin)的重要原因之一。不同的是,哺乳类动物和植物细胞中DNA甲基化修饰的位置及修饰后的生物学功能并不完全相同。在哺乳类细胞内,DNA的甲基化修饰常见于散布在启动子上游的“CpG”二核苷酸位点中的C碱基上。这些位点处的“CpG”中的“C”碱基的第5位可以在DNA甲基转移酶催化下接受来自S-腺苷甲硫氨酸的甲基。未甲基化修饰的CpG被含有“CXXC”结构域的蛋白(CXXC domain-containing proteins)识别,而甲基化修饰的CpGs 则被含甲基结合域(methyl binding domain,MBD)的蛋白识别。植物细胞内的DNA甲基化除发生在CpG位点之外,还能在“CHG”或“CHH”等部位进行,其中“H”可以是A、T、C任一碱基。有意思的是,与哺乳类动物基因中“CpG”散聚在启动子上游的情况不同,植物细胞内的“CpG”很少以“CpG岛”的形式分布于启动子上游。即便如此,植物细胞中DNA的胞嘧啶的甲基化修饰也并不是随机进行的。在植物基因组中,能够被甲基化修饰的“CpG”通常分布在转座子聚集的重复DNA序列、着丝粒重复DNA序列和呈沉默状态的5S或45S rRNA基因组成的重复DNA序列部位。除此之外,植物DNA序列中胞嘧啶的甲基化修饰也出现在受差异调节的启动子和高度表达的基因的蛋白质编码区[4,5]。与动物不同,植物细胞内的CpG甲基化的功能尚未能最终确定,但从其富集在外显子部位来看,可能与mRNA前体的拼接成熟有关。
迄今尚未发现5-meC的“去甲基酶”。而在寻找“5-meC去甲基酶”的过程中却发现了“5-meC”的氧化现象。有关的最新进展表明,DNA中的“5-meC”可被一组名为“10-11”移位“TET”(ten eleven translocation,TET)蛋白分别氧化成5-羟甲基胞嘧啶(5-hydroxymethylcytosine,5-hmeC)、5-甲酸基胞嘧啶(5-formylcytosine,5-fC)和5-羧基胞嘧啶(5-carboxylcytosine,5-caC)。其中,5-hmeC会随DNA复制而丢失,而5-fC和5-caC则会通过DNA胸腺嘧啶糖基化酶(DNA glycosylate,TDG)催化的碱基切除修复去除。由于这些都有助于使“5-meC”重新变为未甲基化的胞嘧啶,因此,5-hmeC、5-fC和5-caC曾一度被认作5-meC去甲基反应的中间体。但是,后续的研究表明,5-hmeC普遍存在于成年生物个体内多种类型的成熟细胞的基因组内,如一些免疫细胞基因组内的5-hmeC约占其碱基总数的万分之五(0.05%),而在浦肯野细胞(purkinje cells)中则要占到其总碱基数的千分之六(0.6%)。并且也发现,包括MeCP2、MBD3和Uhrf2等在内的参与基因转录调控的蛋白均能够识别5-hmeC。因此,5-hmeC更有可能同5-meC一样,是一类与基因表达调控有关的新型胞嘧啶修饰。与此不同,与5-fC和5-caC结合的蛋白则大多为DNA修复蛋白,表明它们才更有可能是5-meC去甲基反应的真正中间体。DNA甲基化修饰程度的改变可见于诸多人类疾病过程中。
2.2 组蛋白翻译后修饰
现今发现的组蛋白翻译后化学修饰主要包括:组蛋白N端无序结构域中赖氨酸残基部位的乙酰基化(acetylation)、甲基化(methylation)、泛素化(ubiquitylation)和小分子泛素蛋白修饰(sumoylation);精氨酸残基部位的甲基化和去氨基(deamination)修饰;脯氨酸的异构化(proline isomerization)和谷氨酸-聚-ADP的核糖基化(glutamate poly-ADP ribosylation)修饰;同时,也包括丝氨酸和苏氨酸残基上的磷酸化修饰[1]。其中,组蛋白特定位点的甲基化/去甲基化、乙酰基化/去乙酰基化修饰属于研究最多的组蛋白翻译后表观遗传修饰类型[1,6]。
通常情况下,组蛋白的乙酰化修饰具有“松开”核小体结构中的核心组蛋白和DNA的相互作用的能力,而去乙酰化和随后进行的甲基化修饰则较为复杂,如H3K9和H3K27位点处的甲基化通常会导致“基因沉寂”,使得该染色质区域呈现异染色质状态;与此不同,发生在H3K4和H3K36的甲基化则通常促进基因转录。上述组蛋白翻译后修饰均需要特异性酶催化,包括已经确定的5大类组蛋白乙酰基转移酶(histone acetyltransferases,HAT)、4大类组蛋白去乙酰基酶(histone deacetylases,HDAC)、丝氨酸或苏氨酸激酶(serine or threonine kinases)、磷酸化酶(phosphatases),以及种类繁多的组蛋白甲基转移酶(histone methyltransferases)和组蛋白去甲基酶(histone demethylases)等[1]。
组蛋白的化学修饰可以直接或间接地影响核小体的稳定性及核小体所在染色质区域的折叠状态,在基因转录、DNA损伤修复、染色质压缩(chromosome condensation)、基因组稳定维护等过程中发挥着重要作用[1]。显然,组蛋白修饰在染色质水平增加了基因表达调控的层级和复杂程度,特别是出现在某些组蛋白N端无序结构域内的氨基酸残基的多价化学修饰,如H3K4、H3K36的二价和三价甲基化修饰(染色质区呈现“活性状态”的标识)以及H3K9m3和H3K27的类似甲基化修饰(染色质呈“沉寂状态”的标识)则更加丰富了表观遗传修饰的语义。与此类似,在单个精氨酸残基上的对称和不对称二价甲基化修饰也体现类似的效应。研究进展表明,包括癌症在内的许多复杂性人类疾病过程中都伴随着组蛋白修饰的异常。
2.3 非编码RNA
与DNA甲基化修饰、组蛋白N端氨基酸残基的多种化学修饰一样,非编码RNA(ncRNAs)对基因表达的调控也是表观遗传修饰的主要手段之一。大量证据表明,数以千计的基因的表达调控受非编码RNA分子的调节。ncRNAs一般可以依据大小和功能分为3种主要类型,包括siRNAs、miRNAs和LncRNAs[7-9]。
2.3.1 siRNAs siRNAs是一大类参与“RNA干扰”(RNA interference,RNAi)的非编码RNA。siRNA广泛参与基因表达调控、病毒防御、转座子活性控制、基因印迹等过程。RNAi则是2003年后基因组时代生物学领域所取得的最激动人心的表观遗传学技术成果之一,已经成为众多分子生物学和生物技术实验室的研究内容。并同时成为继“基因敲除”和“基因突变”之后用于探知未知基因功能的又一重要技术。
但由于特异性问题,利用RNA干扰技术对人类特定疾病的治疗之路显得异常曲折。目前,RNAi 技术还只能应用于人类少数疾病的干预和治疗。
2.3.2 microRNAs 小分子RNA(microRNAs,mi-RN-As)是一类含有20-24个碱基的非编码RNA。它们可以与mRNA的3'或5'非翻译区特异性地结合,并借此抑制蛋白质翻译[1]。当前,已经在人类基因组内确定了2 000多种miRNAs,每一种miRNA均可调控很多基因表达。miRNAs广泛参与生物发育、自稳态(homeostasis)和疾病过程。同时,它们也通过碱基序列特异性影响表观遗传标记的分布模式(distribution patterns)。
小分子非编码RNA也参与RNA沉寂(RNA silencing)过程,负责控制某些内源基因及包括病毒、转入的外源基因或转座子等外源“寄生分子”的活性。小分子RNA参与的“沉寂”体现可移动性,可以在细胞之间(一般包括制造出小分子非编码RNA周围的10-15个细胞范围内移动)进行短程转移,也可以在系统水平上进行长程转移(long-range movement)[10]。小分子RNA的这种可移动性取决于它们在“沉寂”过程中所依赖的碱基序列特异性。与此同时,参与RNA沉寂的RNA分子还可以通过RNA依赖的RNA聚合酶(RNA dependent RNA polymerase,RdRp)转化成新的双链RNA(dsRNA),并经过“Dicer”加工成新的siRNA,使原有的RNAi效应得到进一步放大[10]。miRNAs在包括肿瘤、心血管疾病等重大疾病过程中发挥着重要的作用。
2.3.3 LncRNAs 20多年前已经在哺乳类细胞内发现基于非编码RNA的基因剂量补偿效应(dosage compensation),如人类女性细胞内的X染色体失活现象[11]。一条X染色体失活的转录物XIST就是一条编码自基因间转录的非编码长RNA分子(lncRNA)。XIST 从一条X染色体转录之后与PcGs(polycombgroup complex)复合体中的PRC2结合,引发X染色体上大多数基因的沉寂性关闭。此后的数年间,在不同的生物细胞内发现了数千条长非编码RNA。诸多迹象表明,lncRNA介导的表观遗传学修饰为表观遗传调控构造了另一个关键层级。尽管这些长非编码RNA来源不同,但它们的作用机制却大致相似,很多lncRNAs分子可与染色质修饰和重塑复合体直接作用,向染色质的特定部位引导这些表观遗传“催化剂”[12]。
2.4 组蛋白变体和核小体重塑
众所周知,真核生物细胞核内的DNA与核心组蛋白八聚体形成核小体结构,并和非组蛋白一起形成染色质[1]。核小体是含有多种弱相互作用的稳定的实体结构。核小体结构的稳定性直接影响着基因转录的可能性。因此,对核小体核心组蛋白对应的组蛋白变体,以及源于DNA解旋酶的核小体重塑ATPase调节核小体的稳定性就成了基因能否针对环境、代谢过程和分化指令作出及时的应答的关键。
核小体重塑ATPase常以复合体的形式起作用。一方面,核小体重塑ATPase可以借与ATP的结合和水解进行构象改变,同时,也与核小体中的组蛋白和DNA分子作用。因此,核小体重塑ATPase既可以催化核心组蛋白在DNA上滑动,也可以帮助组蛋白变体置换位于核心组蛋白八聚体中的对应亚基。甚至,核小体重塑ATPase还可以使核小体结构部分或完全解离或间接影响染色质的折叠状态。
需要特别强调的是,组蛋白变体和核小体重塑ATPase的作用几乎可见于基因组代谢的所有过程,不仅参与发育过程中的基因表达模式的建立[13],而且也负责针对环境信号作出基因转录水平的快速应答[14],并能参与程式化的基因组复制[15]。核小体内组蛋白-DNA之间的相互作用因有核小体重塑ATPase的催化呈现可逆性和特异性。例如,有些核小体重塑ATPase主要参与基因转录,有些则特异性地参与DNA损伤、修复和同源重组[1]。
因此,编码核小体重塑ATPase的基因缺陷通常只会对所参与的过程产生影响。例如,与生物发育过程有关的核小体重塑ATPase基因缺陷要么影响胚胎的生存,要么使胚胎出现形态缺陷[13],而负责DNA损伤修复的核小体重塑ATPase基因缺陷(如INO80)则只会影响DNA损伤的修复,引发基因组不稳定和癌症[1,16]。
3 表观遗传修饰的遗传力
法国生物学家拉马克(Jean-Baptiste Lamarck)最早提出“获得性状”(acquired traits)可以“遗传”给后代的理论。但该理论很快让位给达尔文的进化论。表观遗传学作为研究基因-环境互作的一门重要生物学分支,近年来的研究进展表明,许多本来属于表观遗传修饰的“获得性状”可以表现出“跨代遗传”现象。这些发现使人们开始重新对拉马克理论进行审慎的思考。例如,来自植物的表观遗传学研究发现,包括春化作用、盐逆境应答及寒冷适应等生理过程中均伴随着表观遗传修饰有关的性状的“跨代遗传”[17]。
对植物适应寒冷环境(cold acclimation)机制的研究发现,拟南芥(Arabidopsis)细胞内组蛋白去乙酰基酶6(hda6)基因突变体不仅表现出比野生型植株更差的抗霜冻能力,而且对一般意义上的寒冷适应能力也明显下降。因此,HDA6-介导的染色质修饰在拟南芥寒冷适应过程中发挥着关键作用[18]。同样,在高盐胁迫下,拟南芥P5CDH、SRO5基因所编码的siRNAs会得到表达,并直接影响拟南芥的抗盐能力[19]。来自玉米的研究表明,寒冷胁迫可以影响玉米基因组DNA的甲基化修饰水平,寒冷的温度可以诱导玉米根部表达的ZmMI1基因呈低水平甲基化状态[20]。与此类似,经过重金属处理的白苜蓿和工业大麻等也可以诱发各自的根部特定基因位点呈现低甲基化修饰水平[21]。即便如此,并不是所有来自环境诱导的表观遗传标记都体现“跨代遗传”现象,植物和动物的许多起因于环境改变的表观遗传修饰模式的改变可以在重新形成生殖细胞过程中被“抹去”,并不能直接传递到子代个体中。事实上,很多表现出跨代遗传的表观遗传性状常见于对植物的研究,这可能与植物生殖器官几乎无一例外地来自其体细胞分化成的“花”,而动物的生殖器官则与生俱来有关。因此,不难理解,植物的生殖细胞有可能会承载着来自体细胞时期所承受的环境刺激所获得的表观遗传修饰。这些表观遗传修饰一旦逃过减数分裂阶段的“重置”,就有可能被传给后代;当然,植物中这种获得性遗传能力也可能与其“重置”获得性性状不彻底有关。有研究表明,植物并不像动物那样能够通过胚胎发生把来自基因-环境互作的表观遗传性状彻底去除[22]。现在,已经在拟南芥中发现了两个染色质调节因子DDM1 和MOM1与阻止逆境(胁迫)诱导的基因转录改变向子代植株传递有关。人为造成ddm1和 mom1基因缺陷可以使环境所诱导的基因转录特征在后代个体再现[23]。
4 免疫力和重大疾病过程中的表观遗传学修饰改变
过敏性反应、动脉粥样硬化(atherosclerosis)、糖尿病(diabetes)和包括阿兹海默症(alzheimer’s disease,AD)、帕金森(parkinson diseases,PD)、弗里德里希共济失调综合征(friedrich’sataxia)在内的神经退行性等疾病的治疗长期困扰医学界。最近发现,这些疾病过程均存在着表观遗传学修饰紊乱问题。
4.1 肌体免疫力的表观遗传学控制
后生生物的免疫力由天然免疫(innate immune system)和获得性免疫(adaptive immune system)组成。其中,肌体的免疫力有赖于免疫系统中免疫细胞的异质性、多样性及对病原体的反应能力。包括过敏、哮喘和慢性肺梗阻等在内的疾病多与肌体免疫反应异常有关。最近的研究表明,组蛋白乙酰化和甲基化修饰对于巨噬细胞耐受培育起着关键的作用。同样,DNA“CpG”的甲基化修饰也在T调节细胞的发育和功能发挥方面起着关键的作用。有报道表明,包括严重哮喘、吸烟引发的哮喘和肺慢阻(COPD)等呼吸道疾病患者肺部的巨噬细胞和血细胞中I类组蛋白去乙酰基酶HDAC2的表达呈现低水平。对于这类病患使用组蛋白去乙酰基酶抑制剂(HDACi)常可加重炎性基因表达。由于HATs 和 HDACs 还可以修饰包括转录因子在内的其他非组蛋白,并最终改变有关蛋白的活性。因此,利用组蛋白去乙酰基酶(HDAC)抑制剂曲古抑菌素A(trichostatin A)可以缓解试验动物小鼠的过敏性哮喘。在这个过程中,曲古抑菌素A被认为有可能通过对非组蛋白类蛋白的乙酰化而促进了有关细胞死亡。这种情况表明,组蛋白的过度乙酰化修饰与异常的炎性反应有关,而进一步抑制组蛋白去乙酰基酶活性在加剧炎性反应的同时,还可以进一步加剧乙酰基转移酶对组蛋白之外的蛋白的乙酰化修饰,并因此诱发细胞死亡[24]。
4.2 恶性肿瘤过程中的表观遗传修饰缺陷
在参与表观遗传的修饰酶中有几类修饰酶扮演着“癌基因”和“肿瘤抑制基因”的角色[1,25]。例如,人类恶性淋巴瘤细胞内常见 CREBBP、EP300、MLL1、MLL2、PRC2、MMSET和SETD2等 修 饰酶和UTX 去甲基化酶基因突变或功能丧失[6]。约有41% 的囊泡型淋巴瘤患者(follicular lymphoma,FL),39% 的融合型大B细胞淋巴瘤(DLBCL)和18% 复发型B细胞急性淋巴母细胞白血病患者细胞内常见组蛋白乙酰基转移酶CREBBP(CBP)或EP300(p300)的单个等位基因突变,这些基因突变可造成BCL6、p53和组蛋白H3等基因的低水平乙酰化修饰,使得相应基因低水平表达。因此,CREBBP、EP300在上述肿瘤发生过程中扮演着“肿瘤抑制基因”的角色。在包括浆细胞来源在内的多种恶性骨髓瘤细胞中,常见组蛋白H3K36 二甲基化转移酶MMSET(NSD2和WHSC1)的高水平表达,导致基因组范围内H3K36me2水平过高,一般认为,基因组范围内的H3K36me2修饰类型的改变可能会造成局部染色质的“松弛”,并因此活化沉默状态的基因表达,其中可能包括促进浆细胞转化的基因,因此MMSET的高表达扮演着“癌基因”的角色。在T细胞前体急性淋巴母细胞白血病(ETPALL)患者细胞内,可见组蛋白H3K36 三价甲基化转移酶SETD2的缺乏,直接影响H3组蛋白第36位赖氨酸残基的甲基化修饰;而对于恶性B细胞癌患者而言,细胞内丢失H3K27去甲基酶UTX[6],以及一个改变特异性的基因突变和H3K27甲基转移酶EZH2的过表达,致使基因沉寂标记H3K27me3增多。而EZH2甲基转移酶的丢失又常见于急性T细胞白血病(T-ALL)。在B细胞淋巴瘤患者细胞内,H3K4甲基转移酶MLL2的失活可极大降低H3K4me3的水平,而在MLL1-重排型白血病患者细胞内,MLL1融合蛋白则特异性引导甲基转移酶DOT1L 对H3K79进行甲基化修饰,表现出“癌基因”行为。不仅如此,某些肿瘤抑制基因产物也需要借助表观遗传修饰的酶发挥作用,如Rb可以借与I类组蛋白去乙酰基酶的作用,引导它们在特定基因的启动子部位除去原有的乙酰基,并与修饰H3K9me3的组蛋白H3K9三价甲基化转移酶Suv39h1/2作用,促进H3K9me3的形成和异染色质的出现,最终导致该染色质区域内的基因沉默。经过多年研究,表观遗传引发的基因沉默(epigenetic gene silencing)已经被确定为癌细胞发展的重要标志之一。DNA甲基化修饰和组蛋白的去乙酰基化与之密不可分。因此,DNA甲基转移酶(DNA methyltransferase,DNMTs)和组蛋白去乙酰基转移酶(histone deacetylases,HDACs)已经成为癌症表观遗传治疗优选的靶点。
4.3 动脉粥样硬化过程中的表观遗传学改变
动脉粥样硬化是因体内脂质代谢异常引发的心血管疾病[26],在美国、欧洲和日本等发达国家,每年死于动脉粥样硬化的人数接近总死亡人数的一半以上。在我国,动脉粥样硬化的发病率也表现出高发和年轻化特点。
在动脉粥样硬化过程中,高浓度的低密度脂蛋白在血管内膜被氧自由基过氧化为氧化型低密度脂蛋白,引发血管内皮和血管壁慢性炎性反应。其间,炎性细胞进一步释放炎性因子,吸引更多的免疫细胞,最终使血管壁发生进行性损伤,导致血管壁增厚,管腔变窄[27]。
现有的研究表明,DNA甲基化修饰、组蛋白翻译后修饰和非编码RNA的行为都会影响动脉粥样硬化的病程[33]。从患有动脉粥样硬化症患者体内分离出来的DNA总体表现低甲基化水平[28]。但与血管健康维护有关的基因,如超氧化物歧化酶、雌激素受体α和内皮一氧化氮合成酶的血管内物质的基因启动子区却呈现高度甲基化状态,显然,这些“好”的基因的启动子附近的“CpG”高度甲基化与相应基因编码产物表达水平降低有关[29-32]。因此,在治疗中可以将DNA甲基化转移酶作为潜在的靶点来影响体内DNA甲基化水平。
动脉粥样硬化经常出现粥样斑块破裂,斑块破裂涉及到游离于细胞核和细胞质间的I类组蛋白去乙酰基酶HDAC3。这个过程中,HDAC3是唯一表现表达量“上调”的组蛋白去乙酰基酶。研究表明,针对HDAC3活性的抑制剂可以使斑块巨噬细胞转向抗炎症反应并减少脂质的积累。
需要提及的是,一些天然食物组分含有某些组蛋白修饰酶的抑制性物质,如花椰菜中的萝卜硫素就表现出组蛋白去乙酰化转移酶抑制剂的作用[34]。同时,也可以作为抗氧化剂对Nrf2途径间接进行表观遗传修饰的调解,对包括血红素氧合酶-1、NAD(P)H脱氢酶、谷胱甘肽S转移酶、铁蛋白及硫氧还蛋白等一系列抗氧化剂、解毒酶和蛋白质的表达均有影响,而这些物质对于调节炎症紊乱具有作用[34-37]。类似的还有姜黄素[38]和原儿茶醛[39]。其中,对小鼠给予0.4%的姜黄素连续4个月的观察发现,姜黄素可以减少其动脉粥样硬化损伤,并诱导包括细胞黏附、内皮细胞转移等有关的基因的表达[38]。另有报道显示,将姜黄素与萝卜硫素联合使用可治疗胰腺癌。而原儿茶醛通常通过中药用于治疗多种血管性疾病。
4.4 糖尿病过程中的表观遗传学改变
糖尿病是一大类由胰岛素分泌缺陷和/或胰岛素作用障碍引发的以高血糖为特征的糖代谢性紊乱性疾病。临床上常见I型糖尿病和II型糖尿病[40]。I型糖尿病本质上是一种自身免疫性疾病,患者的胰岛β(beta)细胞因受自身免疫细胞的攻击而呈现慢性的炎症状态(称为胰岛炎),这种慢性炎症可以最终完全破坏β细胞,引发胰岛素缺乏[41]。
临床上,I型糖尿病患者需要定期注射胰岛素降低体内血糖水平[42]。最近的研究表明,一种合成的组蛋白仿制品(类似物)药物I-BET151可以在试验动物身上表现出明确的降糖疗效。I-BET151不仅可以有效阻止小鼠的胰岛炎,而且可以对已经发生的胰岛炎具有治疗效果[43]。I-BET151与乙酰化修饰的组蛋白类似,可以模拟乙酰化的组蛋白结构干扰溴结构域蛋白Brd4对乙酰化表观遗传修饰密码的解读,并借此阻止乙酰化对基因的活化[44]。除此之外,JQ1也可以起到Brd4类似物的抑制剂作用,也能够用来阻止Brd4与乙酰化组蛋白的结合[45]。
I-BET151药物还会增加抗炎症物质的表达[46]。Brd4中含有110个氨基酸残基的肽段以正向串接的两个模块BDI 和 BDII组成的溴结构域(bromodomain),经乙酰化修饰的组蛋白可以通过溴结构域得到识别。这种结构也存在于BET蛋白家族的相关蛋白BRD2 和BRD3 中[47]。利用具有高度选择性的人工合成的拮抗剂I-BET或JQ1等阻断溴结构域与乙酰化组蛋白的作用可以阻断BET蛋白与染色质的结合。因此JQ1 和I-BET 可以影响包括IL-12、IL-6在内的众多炎症因子的基因表达。
II型糖尿病常见于成人中,主要是由于机体对胰岛素的抵抗作用所致[48]。已知的II型糖尿病中胰岛素分泌的阻断机制包括由高血糖、高血脂和氧化逆境共同引起的积累损伤。其中,氧化压力和氧自由基被认为会直接影响DNA甲基化和组蛋白的组织,进而影响多种基因的表达[49,50]。
4.5 神经退行性疾病中的表观遗传学
经过数十年的的研究发现,包括阿兹海默症、帕金森疾病、弗里德里希共济失调综合征、脆性X染色体综合征、亨廷顿氏疾病和脊髓小脑共济失调症等神经退行性疾病的发生及发展均与表观遗传学标记的改变有关[51-53]。
4.5.1 阿兹海默症中表观遗传学改变 阿兹海默症(AD),又称老年痴呆症,是一种常见于中老年人群中的神经退行性疾病。该病可随着时间的推移而恶化,目前尚缺乏有效的治疗手段。大多数AD患者受到影响的大脑区域出现胞外淀粉状沉淀物,称为老年斑(SP)和神经元内的磷酸化tau蛋白的积累,成为神经纤维纠缠结(NFT)[54]。
早老素基因的突变可引发早发性痴呆(患者年龄<65)。然而,大部分AD患者(95%-98%)都是迟发性痴呆,是由包括基因、环境和随机的因素等的复杂作用所引起的[55]。在这类AD患者中,基因和环境相互作用有关的表观遗传修饰紊乱在疾病过程中扮演了重要角色[55]。
利用细胞培养和动物模型的研究发现,死于AD的患者脑内出现5-meC和5-hmeC的整体水平降低的现象[56,57],同时,颞叶细胞中H3乙酰基化水平降低[58],组蛋白去乙酰基化转移酶HDAC6和HDAC2的水平升高,这种改变首先会导致tau磷酸化水平的升高[59,60]。因此,在AD动物模型中,使用组蛋白去乙酰基转移酶抑制剂(histone deacetylase inhibitor,HDACi)处理,可表现出疾病的缓解和记忆力不同程度的恢复[61]。此外,在AD患者中,一碳单位代谢受损可直接影响DNA甲基化水平[62]。目前,在AD动物模型中使用广泛性HDACi(表1),如丙戊酸[63]、曲古抑菌素A[64]和丁酸钠[65]等,以及具有特异性的HDACi如MS275[66]和W2[67]均可影响组蛋白修饰状况,有助于缓解试验动物的AD症状[63-69]。
4.5.2 帕金森疾病中表观遗传学改变 帕金森疾病(PD)是仅次于AD的第二大类神经退行性疾病,临床上表现为静止性震颤、僵硬和运动迟缓、姿态不稳[70]和非运动性表现症状,包括认知障碍和睡眠障碍等。病理学上,PD表现为神经黑色素的丢失,包括黑质中的多巴胺能神经元[71]。
尽管使用左旋多巴和多巴胺能治疗方法可以改善PD的一些症状,但目前仍然没有一种有效的方法能阻止PD的发展进程[72]。
大量的研究表明,PD的表观遗传学修饰集中在PD基因的启动子部位甲基化紊乱[73]。其中,作为最早发现的PD基因的SNCA基因[74],其编码α-突触核蛋白。在PD患者的黑质区观察到SNCA基因的甲基化水平降低[75]。此外,α-突触核蛋白的螯合剂DNMT1的水平也呈下降趋势[76]。研究表明,一些HDACi可作为神经保护剂来阻止由α-突触核蛋白介导的神经元毒性,如丙戊酸[77]、丁酸钠[78-80]、vorinostat[81]和AK-1[82]等。
以动物为研究模型的试验表明,对暴露在除草剂中的小鼠的黑质神经元进行分析,其α-突触核蛋白转移至细胞核并与组蛋白结合[83]。而在对果蝇的研究中发现,α-突触核蛋白介导细胞核中的神经元毒性,并直接与H3结合且抑制组蛋白乙酰化,α-突触核蛋白的毒性可使用丁酸钠或vorinostat降解[81]。组蛋白乙酰化转移酶2的选择性抑制作用可在由α-突触核蛋白介导的毒性中发挥作用[82]。此外,大量的研究表明,HDACi对神经元上诱导的神经毒素损害有保护作用[84]。
4.5.3 三核苷酸扩增有关的神经-肌肉退行性疾病与表观遗传学 亨廷顿疾病、脊髓小脑共剂失调、强制性肌营养不良、弗里德里希共剂失调、脆性X染色体及其相关的疾病是长期困扰国际医学界的与三核苷酸重复序列扩增相关的疾病(表2)[1,85-87]。最近的研究表明,这些疾病过程中伴随着表观遗传学修饰的方式改变和重复序列重复数目的增加。迄今医学界对上述疾病的干预和治疗束手无策。人们普遍寄希望于利用可校正表观遗传修饰化合物对这类重大疾病进行有效的干预或治疗[88,89]。
4.5.3.1 弗里德里希共济失调(Friedreichs ataxia,FRDA)与表观遗传学 FRDA是一种常染色体隐性神经退行性线粒体紊乱疾病,主要由在FXN基因上的第一个内含子上的GAA重复序列扩增引起[86,87,90]。正常人体内的GAA重复序列数不超过43个重复单位,而患者体内的GAA重复序列数多达1 700(44-1 700)[91]。
GAA重复序列扩增导致线粒体蛋白Frataxin的表达量降低[92],故使用能够诱导Frataxin表达的药物提高Frataxin的表达量对于治疗该病有益。最近的研究表明,FXN基因沉默与表观遗传学的控制有着密切的关系。在FRDA患者中,DNA甲基化水平升高,因此,有人提出使用去甲基化药物激活沉默的FXN基因的设想[93]。候选的去甲基化药物可为核苷类似物去甲基化抑制剂和非核苷类似物去甲基化抑制剂[94]。然而,目前尚缺乏利用DNA去甲基化药物作为治疗FRDA方法的报道。主要是尚不十分明确DNA甲基化与FXN基因沉默的关系。
理论上讲,可使用包括丁酸盐、苯甲酰胺复合物BML-210和氨基苯化合物[96]等HDAC抑制剂增加组蛋白、转录因子和其他转录相关蛋白的乙酰化水平来影响转录水平[95]。而最近的合成药物109则可能是最有希望用于治疗FRDA的药物,目前已进入临床I期验证[97],
除此之外,抗原RNAs(agRNAs)也有望用于FRDA的表观遗传学治疗。agRNAs是一些19 bp的RNAs,可以靶向作用于FXN基因的启动子区[98]。因此,有望使用agRNAs靶向激活FXN基因表达。
4.5.3.2 脆性X染色体综合征(FXS) 脆性染色体综合征(Fragile X Syndrome,FXS)是由于X染色体上一段三核苷酸重复序列扩增引发的综合征[1,85]。FXS患者细胞内FMR1基因的5' UTR区的CGG重复序列扩增数大于200个重复单位之后即关闭FMR1基因的表达,造成相应基因产物缺失。病患个体早期神经系统发育会因此受累,出现面部异形、行为障碍和认知困难[99,100]。FXS在男性中的发病率明显高于女性,分别为万分之四(0.04%)和万分之二点五(0.025%)[99,101],这可能与女性体内含有一个非扩增性的FMR1等位基因有关[99]。
现在认为脆性X染色体综合征患者体内FMR1基因沉默与CGG重复序列区以及其上游的CpG岛的异常甲基化有关[102,103]。研究发现,绝大多数患
者CGG重复序列区的“CpG岛”以及其上游的启动子区都呈高度甲基化状态[103-105]。因此,这些表观遗传学标记将会被用作一种新型的从控制上区分男性和女性患者的DNA诊断方式[106]。

表1 表观遗传修饰酶抑制剂及拮抗剂
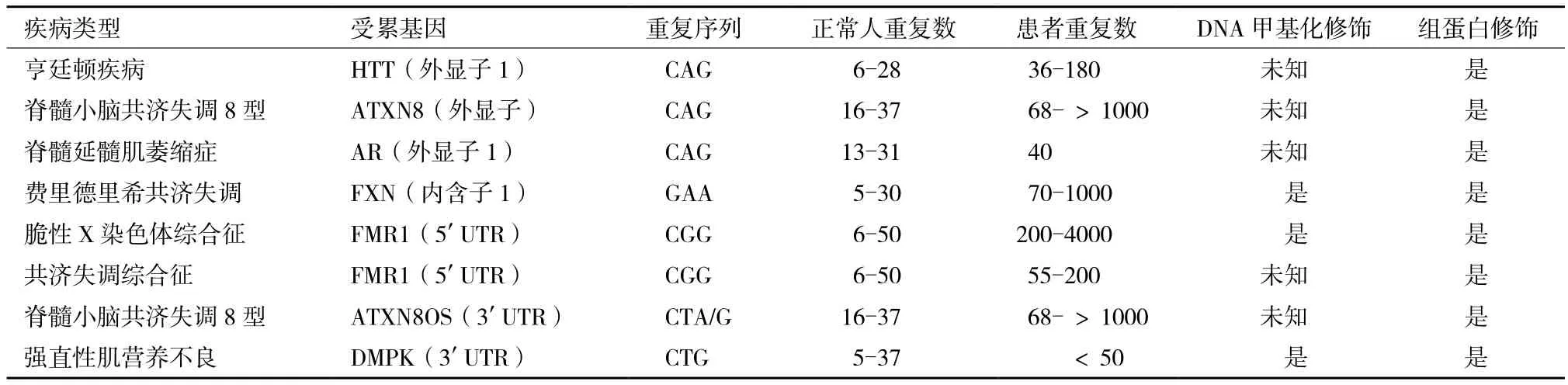
表2 目前已知的神经退行性疾病类型与相关的表观遗传学修饰
5 基于表观遗传学机制的生物技术和药物的研发
在过去的20年里,围绕DNA甲基化修饰、组蛋白翻译后修饰过程中催化剂展开的抑制剂筛查的医药生物技术研发取得了长足的进展。目前,已经得到了一大批有望用于肿瘤和癌症表观遗传治疗的修饰酶抑制剂(表1)。与此同时,基于小分子非编码RNA表观遗传作用机制的RNA干扰技术也日臻完善,并已被用于对未知基因功能的探查的基础研究和对诸多癌症的干预和治疗实践。
大量的证据表明,癌症发生过程中伴随着表观遗传修饰紊乱,导致癌细胞内许多癌基因的高表达和抑癌基因的沉寂。利用所筛选出的DNA甲基化酶抑制剂和组蛋白去乙酰基酶抑制剂已经表明可以有效地恢复某些肿瘤细胞内的“肿瘤抑制基因”表达的抑制和“癌基因”的高表达(表1)。当前,常规的抗肿瘤药物都无一例外地表现出高度低效问题,而使用表观遗传药物或同时辅助常规抗肿瘤药物则可以明显优于单独使用常规药物的疗效。现在,利用DNA甲基化转移酶抑制剂和组蛋白去乙酰基酶抑制剂已经在包括乳腺癌、肺癌、宫颈癌、直肠癌及白血病等癌症治疗和干预实践中取得了成效(表1)。
相较于基因突变引起的遗传缺陷而言,基因的表观遗传修饰紊乱有关的疾病具有可逆的特点,因此,有望利用表观遗传药物对包括癌症等疾病在内的疾病细胞实现“逆转”,使它们重新恢复为机能正常的细胞或被特异性地去除。
近几年的研究也表明,在长期困扰医药界的阿兹海默症、帕金森、弗里德里希共济失调综合征等神经退行性疾病的发生过程中也伴随着表观遗传修饰紊乱问题。因此,有望利用能够透过“血脑屏障”的表观遗传修饰酶小分子抑制剂药物实现对这些长期困扰医药界的重大疾病实现有效的干预和治疗。
简言之,当前基于表观遗传机制的生物医药技术的研发已经进入到一个新的发展时期。从过去主要针对肿瘤和癌症的干预及治疗药物的研发扩展成针对包括心脑血管、糖尿病、神经退行性疾病等更多疾病的干预和治疗的表观遗传生物医药技术研发。在过去的10多年里,利用针对癌细胞内的DNA甲基化和组蛋白乙酰化修饰酶类等靶点的表观遗传药物已经在血液癌症治疗方面取得了明显成效,现在,对于针对其他癌症、心血管疾病、代谢性疾病和神经退行性疾病的更具特异性的“第二代”表观遗传药物逐渐呈现出如火如荼的研发势头(表1)。
概言之,基于表观遗传机制的生物医药技术的研发将会围绕以下3个主要方面展开:(1)针对疾病过程中的DNA甲基化和组蛋白甲基化修饰的改变,研发更具特异性和针对性的DNA和组蛋白去甲基化酶抑制剂和甲基激活剂,通过使用这些药物“校正”患者体内DNA甲基化水平和组蛋白甲基化修饰,使其恢复到正常人的甲基化水平,从而达到治疗目的;(2)使用更具针对性和特异性的去乙酰基转移酶抑制剂(HDACi)影响疾病过程中的低水平组蛋白乙酰基化修饰水平,激活被表观遗传修饰沉默的基因的表达水平;(3)利用靶向输送技术输送更具特异性的靶向非编码RNA至病灶部位,进一步提高非编码RNA干扰的特异性和有效性,通过影响与疾病过程密切相关的蛋白基因的表达实现对疾病的干预或治疗。
6 结语
自20世纪90年代以来,表观遗传学及基于表观遗传机制的生物医药技术的发展已经成为举世瞩目的生物学热点。在短短20年里,DNA甲基化修饰、组蛋白翻译后修饰和非编码RNA等表观遗传基础研究已经取得了长足进展。特别是围绕癌症、心血管疾病和神经退行性疾病等重大疾病过程中的表观遗传修饰异常的研究,不仅有助于理解生物的发育、分化、自稳态维持、基因-环境互作等生物学基础问题,而且也有助于快速研发出更具针对性,更加有效的疾病治疗药物和技术方法。在过去的几年里,一些基于癌症表观遗传机制改变的药物已经获批用于临床治疗。相较而言,虽然针对心脑血管、神经退行性疾病的表观遗传药物研究起步稍晚,但发展势头强劲。就当前的总体趋势来看,基于表观遗传修饰机制的药物和治疗技术普遍存在副作用大,特异性低的问题。因此,如何有效利用现有分子生物学和结构生物学的技术手段,进一步深入解析参与表观遗传修饰的生物大分子和酶类的结构与功能,更有效确定更具特异性的表观遗传药物已经成为当前亟需解决的问题。尽管存在着这样或那样的问题,但我们完全有理由相信,随着表观遗传修饰机制过程研究的日益深入,基于表观遗传机制的疾病治疗技术和药物研发终将会取得突破性的进展。
[1]潘学峰. 基因疾病的分子生物学[M]. 北京:化学工业出版社, 2014:1-450.
[2]Barlow DP, Bartolomei MS. Genomic imprinting in mammals[M]. Cold Spring Harb Perspect Biol, 2014, 6:a018382.
[3]Kriaucionis S, Tahiliani M. Expanding the epigenetic landscape:novel modifications of cytosine in genomic DNA[M]. Cold Spring Harb Perspect Biol, 2014, 6:a018630.
[4]Zilberman D, Gehring M, Tran RK, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription[J]. Nat Genet, 2007, 39: 61-69.
[5]Feng S, Cokus SJ, Zhang X, et al. Conservation and divergence of methylation patterning in plants and animals[J]. Proc Natl Acad Sci, 2010, 107:8689-8694.
[6]Mar BG, Bullinger L, Basu E, et al. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia[J]. Leukemia, 2012, 26:1881-1883.
[7]Mercer TR, Dinger ME, Mattick JS. Longnon-coding RNAs:insights into functions[J]. Nat Rev Genet, 2009, 10:155-159.
[8]Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs[J]. Cell, 2009, 136:629-641.
[9]Kaikkonen MU, Lam MT, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics[J]. Cardiovasc Res, 2011, 90:430-440.
[10]Melnyk CW, Molnar A, Bassett A, Baulcombe DC. Mobile 24 nt small RNAs direct transcriptional gene silencing in the root meristems of Arabidopsis thaliana[J]. Curr Biol, 2011, 21:1678-1683.
[11]Brockdorff N, Turner BM. Dosage compensation in mammals[M]. Cold Spring Harb Perspect Biol, 2014:a019406.
[12]Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine[J]. Science, 2008, 319:1787-1789.
[13]Chioda M, Becker PB. Soft skills turned into hard facts:nucleosome remodelling at developmental switches[J]. Heredity, 2010, 105:71-79.
[14]Vicent GP, Nacht AS, Zaurin R, et al. Minireview:role of kinases and chromatin remodeling in progesterone signaling to chromatin[J]. Mol Endocrinol, 2010, 24:2088-2098.
[15]Morettini S, Podhraski V, Lusser A. ATP-dependent chromatin remodeling enzymes and their various roles in cell cycle control[J]. Front Biosci, 2008, 13:5522-5532.
[16]Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling:genetics, genomics and mechanisms[J]. Cell Res, 2011, 21:396-420.
[17]Baulcombe DC, Dean C. Epigenetic regulation in plant responses to the environment[M]. Cold Spring Harb Perspect Biol, 2014, 6:a019471.
[18]To TK, Nakaminami K, Kim JM, et al. Arabidopsis HDA6 is required for freezing tolerance[J]. Biochem Biophys ResCommun, 2011, 406:414-419.
[19]Borsani O, Zhu J, Verslues PE, et al. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis[J]. Cell, 2005, 123:1279-1291.
[20]Steward N, Ito M, Yamaguchi Y, et al. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress[J]. J Biol Chem, 2002, 277:37741-37746.
[21]Aina R, Sgorbati S, Santagostino A, et al. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp[J]. Physiol Plant, 2004, 121:472-480.
[22]Gutierrez-Marcos JF, Dickinson HG. Epigenetic reprogramming in plant reproductive lineages[J]. Plant Cell Physiol, 2012, 53:817-823.
[23] Iwasaki M, Paszkowski J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states[J]. Proc Natl Acad Sci USA, 2014, 111:8547-8552.
[24] Oh ET, Park MT, Choi BH, et al. Novel histone deacetylase inhibitor CG200745 induces clonogenic cell death by modulating acetylation of p53 in cancer cells[J]. Investigational New Drugs, 2010, 30(2):435-442.
[25] Busslinger M, Tarakhovsky A. Epigenetic control of immunity[M]. Cold Spring Harb Perspect Biol, 2014, 6:a019307.
[26] Weber C, Noels H. Atherosclerosis:current pathogenesis and therapeutic options[J]. Nat Med, 2011, 17:1410-1422.
[27] Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis[J]. Cell, 2011, 145:341-355.
[28]Sharma P, Kumar J, Garg G, et al. Detection of altered global DNA methylation in coronary artery disease patients[J]. DNA Cell Biol, 2008, 27:357-365.
[29]Castro R, Rivera I, Blom HJ, et al. Homocysteine metabolism, hyperhomocysteine mia and vascular disease:an overview[J]. J Inherit Metab Dis, 2006, 29:3-20.
[30] Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis:transition from theory to practice[J]. Circ J, 2010, 74:213-220.
[31] Jia L, Zhu L, Wang JZ, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronaryartery disease[J]. Atherosclerosis, 2013, 228:346-352.
[32] Zaina S, Heyn H, Carmona FJ, et al. A DNA methylation map of human Atherosclerosis[J]. Circ Cardiovasc Genet, 2014, 7(5):692-700.
[33] Mikaela MB, Ross TM, Anthony WR. Epigenetic modulation in the treatment of atherosclerotic disease[J]. Fronries in Genetics, 2014, 364:1-7.
[34]Myzak MC, Tong P, Dashwood WM, et al. Sulf oraphanere tards the growth of human PC-3xenografts and inhibits HDAC activity in human subjects[J]. Exp Biol Med(Maywood), 2007, 232:227-234.
[35]Elbarbry F, Elrody N. Potential health benefits of sulforaphane:a review of the experimental, clinical, and epidemiological evidences and underlying mechanisms[J]. J Med Plants Res, 2011, 5:473-484.
[36]Bai Y, Cui W, Xin Y, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulaton of Nrf2 expression and transcription activation[J]. J Mol Cell Cardiol, 2013, 57:82-95.
[37]Zhang C, Su ZY, Khor TO, et al. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMPC1 cells through epigenetic regulation[J]. Biochem Pharmacol, 2013, 85:1398-1404.
[38]Coban D, Milenkovic D, Chanet A, et al. Dietary curcumin inhibits atherosclerosis by affecting the expression of genes involved in leukocyte adhesion and transendothelial migration[J]. Mol Nutr Food Res, 2012, 56, 1270-1281.
[39]Moon CY, Ku CR, Cho YH, et al. Protocatechuic aldehyde inhibits migration and proliferation of vascular smooth muscle cells and intravascular thrombosis[J]. Biochem Biophys Res Commun, 2012, 423:116-121.
[40]Dean L, McEntyre J. The Genetic Landscape of diabetes[M]. Bethesda(MD):National Center for Biotechnology Information 9US0, 2004:1-135.
[41]Aathira R, Jain V. Advances in management of type 1 diabetes mellitus[J]. World J Diabetes, 2014, 5(5):689-696.
[42]Singh AK, Sinha B. Advances in basal insulin therapy:lessons from current evidence[J]. J Indian Med Assoc, 2013, 111(11):735-736, 738-742.
[43]Fu W, Farache J, Clardy SM, et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and β cells[J]. ELife, 2014, 3:e04631.
[44] Huang B, Yang XD, Zhou MM, et al. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA[J]. Molecular and Cellular Biology, 2009, 29:1375-1387.
[45]Zhang G, Liu R, Zhong Y, et al. Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition[J]. Journal of Biological Chemistry, 2012, 287:28840-28851.
[46]Zou Z, Huang B, Wu X, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA[J]. Oncogene, 2014, 33:2395-2404.
[47]Zeng L, Zhou MM. Bromodomain:An acetyl-lysine binding domain[J]. FEBS Lett, 2002, 513:124-128.
[48]Brietake SA. Oral antihyperglycemic treatment options for type 2 diabetes mellitus[J]. Med Clin North Am, 2015, 99(1):87-106.
[49]Wallace TM, Matthews DR. Coefficient of failure:a methodology for examining longitudinal beta-cell function in type 2 diabetes[J]. Diabet Med, 2002, 19:465-469.
[50]Robertson RP, Harmon J, Tran PO, et al. Glucose toxicity in betacells:type 2 diabetes, good radicals gone bad, and the glutathione connection[J]. Diabetes, 2003, 52:581-587.
[51]Coppedè F. Advances in the genetics and epigenetics of neurodegenerative diseases[J]. Epigenetics Neurodegener Dis, 2014, 1:3-31.
[52]Mirkin SM. Expandable DNA repeats and human disease[J]. Nature, 2007, 447:932-940.
[53]Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo[J]. Cell Res, 2008, 18:198-213.
[54]Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease[J]. Biochem Pharmacol, 2014, 88:508-516.
[55]Coppedè F. Advances in the genetics and epigenetics of neurodegenerative diseases[J]. Epigenetics Neurodegener Dis, 2014, 1:3-31.
[56]Mastroeni D, Grover A, Delvaux E, et al. Epigenetic changes in Alzheimer’s disease:decrements in DNA methylation[J]. Neurobiol. Aging, 2010, 31:2025-2037.
[57]Chouliaras L, Mastroeni D, Delvaux E, et al. Consistent decrease in global DNA methylation and hydroxy methylation in the hippocampus of Alzheimer’s disease patients[J]. Neurobiol Aging, 2013, 34:2091-2099.
[58] Zhang K, Schrag M, Crofton A, et al. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease[J]. Proteomics, 2012, 12:1261-1268.
[59]Ding H, Dolan PJ, Johnson GV. Histone deacetylase 6 interacts with the microtubule-associated proteintau[J]. J Neurochem, 2008, 106:2119-2130.
[60]Gräff J, Rei D, Guan JS, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain[J]. Nature, 2010, 483:222-226.
[61]Karagiannis TC, Ververis K. Potential of chromatin modifying compounds for the treatment of Alzheimer’s disease[J]. Pathobiol Aging Age Relat, 2012, 2:1-12.
[62]Coppedè F. One-carbon metabolism and Alzheimer’s disease:focuson epigenetics[J]. Curr Genomics, 2010, 11:246-260.
[63]Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease[J]. Neuropsychopharmacology, 2010, 35:870-880.
[64] Francis YI, Fà M, Ashraf H, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease[J]. J Alzheimers Dis, 2009, 18:131-139.
[65] Fischer A, Sananbenesi F, Wang X, et al. Recovery of learning and memory after neuronal loss is associated with chromatin remodeling[J]. Nature, 2007, 447:178-182.
[66] Zhang Z, Schluesener YHJ. Oralad ministration of histone deacetylase inhibitor MS-275 ameliorates neuro inflammation and cerebralamy loidosis and improves behavior in a mouse model[J]. J Neuropathol Exp Neurol, 2013, 72:178-185.
[67] Sung YM, Lee T, Yoon H, et al. Mercaptoacetamide-based classII HDAC inhibitor owers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease[J]. Exp Neurol, 2013, 239:192-201.
[68] Govindarajan N, Agis-Balboa RC, Walter J, et al. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administer edatan advanced stage of disease progression[J]. J Alzheimers Dis, 2011, 26:187-197.
[69] Ricobaraza A, Cuadrado-Tejedor M, Marco S, et al. Phenylbutyraterescues dendritic spine loss associated with memory deficits in amouse model of Alzheimer disease[J]. Hippocampus, 2010, 22:1040-1050.
[70] Mak MK, Cole JH. Movement dysfunction in patients with Parkins-on’s disease:a literature review[J]. Aust J Physiother, 1991, 37(1):7-17.
[71] Devos D, Lejeune S, Cormier-Dequaire F, et al. Dopa-decarboxylase gene polymorphisms affect the motor response to L-dopa in Parkinson's disease[J]. Parkinsonism Relat Disord, 2014, 20(2):170-175.
[72]Thomas B, Beal MF. Molecular insights into Parkinson’s disease[J]. F1000 Med Rep, 2011, 3:7.
[73]Coppedè F. Genetics and epigenetics of Parkinson’s disease[J]. Scientific World Journal, 2012, 2010:1-12.
[74]Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’brains[J]. J Neurosci, 2010, 30:6355-6359.
[75]Matsumoto L, Takuma H, Tamaoka A, et al. CpG demethylation enhances alpha synuclein expression and affects the pathogenesis of Parkinson’s disease[J]. PLoS One, 2010, 5:e15522.
[76]Desplats P, Spencer B, Coffee E, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus:anovel mechanism for epigenetic alterations in Lewy body diseases[J]. J Biol Chem, 2011, 286:9031-9037.
[77]Monti B, Gatta V, Piretti F, et al. Valproic acid is neuroprotective in the rotenonerat model of Parkinson’s disease:involvement of α-synuclein[J]. Neurotox Res, 2010, 17:130-141.
[78]Zhou W, Bercury K, Cummiskey J, et al. Phenylbutyrateupregulates the DJ-1protein and protects neurons in cell culture and in animal models of Parkinson’s disease[J]. J Biol Chem, 2011, 286:14941-14951.
[79]Rane P, Shields J, Heffernan M, et al. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in premotorstage PD[J]. Neuropharmacology, 2012, 62:2409-2412.
[80]St Laurent R, O’Brien LM, Ahmad ST. Sodium butyrate improves locomotor impairment and early mortality in arotenone-induced Drosophila model of Parkinson’s disease[J]. Neuroscience, 2013, 246:382-390.
[81]Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity[J]. Hum Mol Genet, 2006, 15:3012-3023.
[82]Outeiro TF, Kontopoulos E, Altmann SM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein mediated toxicity in models of Parkinson’s disease[J]. Science, 2007, 317:516-519.
[83]Goers J, Manning-Bog AB, McCormack AL, et al. Nuclear localization of alpha-synuclein and its interaction with histones[J]. Biochemistry, 2003, 42:8465-8471.
[84]Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase:therapeutic potential in Parkinson’s disease?[J]. Pharmacol Ther, 2013, 140:34-52.
[85]Pan XF. Mechanism of trinucleotide repeats instabilities:the necessities of repeat non-B secondary structure for-mation and the roles of cellular trans-acting factors[J]. Acta Genet Sin, 2006, 33(1):1-11.
[86]丁云峰, 潘学峰. 三核苷酸重复序列(GAA Trc)n扩增的分子机制研究现状[J]. 国际遗传学杂志, 2009, 32(6):412-415.
[87]Pan XF, Ding YF, Shi LF. The roles of SbcCD and RNaseE in the transcription of GAA TTC repeats in Escherichia coli[J]. DNA Repair(Amst), 2009, 8(11):1321-1327.
[88]Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase:therapeutic potential in Parkinson’s disease?[J]Pharmacol Ther, 2013, 140:34-52.
[89]Karagiannis TC, Ververis K. Potential of chromatin modifying compounds for the treatment of Alzheimer’s disease[J]. Pathobiol Aging Age Relat, 2012, 2:1-22.
[90]Campuzano V, Montermini L, Molto MD, et al. Friedreich’s ataxia:autosomal recessive disease caused by an intronic GAA triplet repeat expansion[J]. Science, 1996, 271:1423-1427.
[91]Pandolfo M. Friedreich ataxia[J]. Arch Neurol, 2008, 65:1296-1303.
[92]Campuzano V, Montermini L, Lutz Y, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes[J]. Hum Mol Genet, 1997, 6:1771-1780.
[93]Sandi C, Sandi M, Anjomani Virmouni S, et al. Epigenetic-based therapies for Friedreich’s ataxia[J]. Frontiers in Genetics, 2014, 5(165):1-12.
[94]Jain N, Rossi A, Garcia-Manero G. Epigenetic therapy of leukemia:an update[J]. Int J Biochem Cell Biol, 2009, 41:72-80.
[95]Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders[J]. Nat Rev Neurosci, 2006, 7:784-796.
[96]Herman D, Jenssen K, Burnett R, et al. Histone deacetylaseinhibitors reverse gene silencing in Friedreich’s ataxia[J]. Nat Chem Biol, 2006, 2:551-558.
[97]Soragni E, Xu C, Plastere HL, et al. Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for Friedreich ataxia[J]. J Child Neurol, 2012, 27:1164-1173.
[98]Watts JK, Yu D, Charisse K, et al. Effect of chemical modifications on modulation of gene expression by duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters[J]. Nucleic Acids Res, 2010, 38:5242-5259.
[99] Gallagher A, Hallahan B. Fragile X-associated disorders:a clinical overview[J]. J Neurol, 2010, 259:401-413.
[100] Loomis EW, Eid JS, Peluso P, et al. Sequencing the unsequenceable:expanded CGG-repeat alleles of the fragile X gene[J]. Genome Res, 2013, 23:121-128.
[101] Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA[J]. Am J Hum Genet, 2009, 85:503-514.
[102] Oberle I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome[J]. Science, 1991, 252:1097-1102.
[103] Pieretti M, Zhang FP, Fu YH, et al. Absence of expression of the FMR-1 gene in fragile X syndrome[J]. Cell, 191, 66:817-822.
[104] Luo S, Robinson JC, Reiss AL, et al. DNA methylation of the fragile X locus in somatic and germ cells during fetal development:relevance to the fragile X syndrome and X inactivation[J]. Somat Cell Mol Genet, 1993, 19:393-404.
[105] Hansen RS, Gartler SM, Scott CR, et al. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene[J]. Hum Mol Genet, 1992, 1:571-578.
[106] Godler DE, Tassone F, Loesch DZ, et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio[J]. Hum Mol Genet, 2010, 19:1618-1632.
[107] Fischer A. Targeting histone-modifications in Alzheimer’s disease. what is the evidence that this is a promising therapeutic avenue?[J]. Neuropharmacology, 2014, 80:95-102.
The Developments of Epigenetics and Epigenetics-based Modern Biomedicine and Pharmaceutics
Jiang Nan1Pan Xuefeng1,2
(1. School of Life Science,Beijing Institute of Technology,Beijing 100081;2. School of Basic Medicine,Hebei University,Baoding 071002)
Epigenetics and epigenetic mechanism-based biomedical technologies have emerged as important research fields in post-genomic area. In-depth investigation on epigenetic alterations of DNA methylation, histone modifications, non-coding RNAs, etc. in tumorigenesis, cardiovascular disease, diabetes, and neurodegenerative diseases associated with the elderly population is not only beneficial to the understanding of the molecular pathogenesis of the diseases, but also useful in exploring effective treatments based upon epigenetic mechanisms. This review first briefly introduces epigenetic modification mechanisms, then summarizes current research progress on abnormal epigenetic modifications in the disease conditions, and also discusses the relevant biomedical technology.
epigenetics;DNA methylation;histone post-translational modification;non-coding RNA;human diseases
10.13560/j.cnki.biotech.bull.1985.2015.03.005
2015-02-02
北京市自然科学基金项目(5132014),北京理工大学自然科学基础基金项目(3160012211215)
姜楠,硕士研究生,研究方向:分子生物学;E-mail:nanjiang_10@126.com
潘学峰,博士,教授,研究方向:分子生物学;E-mail:xuefengpancam@aliyun.com
