先天性肾病综合征儿童的NPHS1基因突变分析
2015-06-28付荣吴清岩徐纪香缑梦帆刘娟同少峰刘雪杰郭辉和俊杰
付荣,吴清岩,徐纪香,缑梦帆,刘娟,同少峰,刘雪杰,郭辉,和俊杰
先天性肾病综合征儿童的NPHS1基因突变分析
付荣,吴清岩,徐纪香,缑梦帆,刘娟,同少峰,刘雪杰,郭辉,和俊杰
目的分析先天性肾病综合征(CNS)患儿的NPHS1基因突变及其特点。方法研究对象为1例中国中部地区汉族CNS患儿及其父母,对照人群为50例尿检正常的汉族成年人。取所有研究对象外周静脉血3ml,提取基因组DNA,PCR扩增NPHS1和NPHS2基因全部外显子及其周围的部分内含子序列,对PCR产物进行直接DNA序列测定。结果在CNS患儿中未检出NPHS2基因突变,检测出NPHS1基因的G928A(D310N)和IVS11+1 G>A 2个杂合突变。患儿母亲尿检正常,基因检测显示NPHS1的G928A(D310N)杂合突变,没有IVS11+1 G>A杂合突变;患儿父亲尿检也正常,基因检测显示NPHS1的IVS11+1 G>A杂合突变,而没有G928A(D310N)杂合突变。在50例对照人群中未发现NPHS1的G928A(D310N)和(或)IVS11+1 G>A突变。结论1例散发性CNS患儿中检测到NPHS1基因的G928A(D310N)和IVS11+1 G>A 2个杂合突变,且IVS11+1 G>A为NPHS1基因剪接位点新突变。CNS患儿应进行NPHS1基因突变分析。
肾病综合征;基因,NPHS1;突变
先天性肾病综合征(congenital nephrotic syndrome,CNS)通常在出生后3个月内发病,临床表现为肾病综合征或大量蛋白尿[1]。CNS具有遗传异质性,大部分患儿起病早、病情重,对糖皮质激素耐药,肾功能呈进行性减退,预后极差;但也有部分患儿临床症状较轻。近年来研究表明,大部分CNS是由于编码肾小球滤过屏障蛋白的基因突变所致,如NPHS1和NPHS2等[2-7]。NPHS1定位于人类染色体19q13.1,是先天性芬兰型肾病(congenital nephrotic syndrome,Finnish type,CNF)的致病基因[1],其编码的蛋白nephrin是第一个被定位于肾小球足细胞裂孔隔膜(SD)上的分子,已被证实是维持肾小球正常滤过功能的关键分子之一。NPHS1基因突变在芬兰CNS儿童中检出率约为98%,在芬兰以外地区的CNS患儿中检出率为39%~80%。国外的研究表明,NPHS1基因突变是CNS较常见的原因,其基因型与表型的关系也成为目前的研究热点之一[2]。目前我国对CNS患者致病基因及其突变的研究还很少。本研究采用聚合酶链反应(PCR)和DNA直接测序的方法,对1例中国中部地区汉族散发性CNS患儿进行NPHS1基因分析,通过复习国内外研究结果,分析其基因型与表型之间的关系,旨在探讨和研究CNS的发病机制。
1 资料与方法
1.1 临床资料 患儿,男,2个月17d。第1胎,第1产,足月顺产,胎盘不详,出生体重2.5kg,生后无窒息。生后20d出现腹胀,诊断为先天性巨结肠。到另一家医院就诊时发现患儿水肿,查尿常规示蛋白(未做蛋白定量)。入院前2周出现发作性抽搐,水肿逐渐加重,为进一步诊治来我院,拟诊“肾病综合征(先天性)”,收住入院治疗。入院体检:体重4000g,呼吸平稳,面部及四肢轻至中度水肿,心肺听诊正常,腹部膨隆,肝脏右肋下可触及2cm,脾脏肋下未触及,神经系统未见异常。尿常规:尿蛋白,24h尿蛋白定量因留取标本困难未查;血生化:总蛋白16.9g/L,白蛋白10.9g/ L,尿素氮1.16mmol/L,肌酐22.1μmol/L,总胆固醇6.66mmol/L;抗巨细胞抗体、弓形虫抗体、风疹病毒抗体、单纯疱疹病毒抗体、支原体抗体、衣原体抗体、梅毒抗体及人免疫缺陷病毒抗体均阴性;甲、乙和丙型肝炎病毒抗体均阴性;甲状腺功能检查正常;血液串联质谱未提示明显异常,尿有机酸测定未见异常。心脏彩超心内结果正常。肾脏彩超示:左肾58mm×34mm,右肾59mm×35mm,回声增强。诊断为先天性肾病(CNS)。因家长不同意,未做肾穿刺活检。未用激素治疗,仅给予对症及口服卡托普利治疗。出院后随访至目前,患儿身高85cm,体重13kg,尿蛋白,肾功能正常。
患儿父亲24岁,母亲24岁,均身体健康,尿常规正常,家族中其他成员均无肾脏病史。以50例尿检正常的汉族成年志愿者作为对照人群,男39例,女11例,年龄18~23岁。
1.2 研究方法
1.2.1 基因组DNA的提取 在获得伦理委员会批准和征得患儿父母及对照人群知情同意后,采集上述研究对象的外周血标本,应用EZNATM SE Blood DNA试剂盒(美国Omega公司)抽提基因组DNA。
1.2.2 基因突变分析 采用文献报道[8-9]的引物序列和实验条件,PCR分别扩增NPHS1和NPHS2基因全部外显子及其周围的部分内含子。引物由上海生工生物技术有限责任公司合成。PCR产物直接测序:以正、反向引物为测序引物,应用Perkin-Elmer公司的ABI377测序仪(上海生工生物工程技术有限公司)进行DNA序列测定。对测序异常的片段重新进行PCR扩增,再次正向、反向测序,以验证结果的可靠性。
1.2.3 突变序列的命名 根据从NCBI的Nucleotide数据库中获得的NPHS1基因组序列(GeneBank:NT_011109.16)、mRNA序列(GeneBank:N M_0 0 4 6 4 6)、蛋白质序列(G e n e B a n k:NP_004637)和人类DNA序列变异命名建议命名为NPHS1基因突变。
1.2.4 突变位点查新 跟踪检索NCBI所属的PubMed文献数据库、单核苷酸多态性(simple nucleotide polymorphism,SNP)数据库(SNP database,dbSNP)公布的NPHS1基因变异,将本研究中经DNA测序证实的基因变异与上述变异进行比较,以确定其是否为新发现的NPHS1基因变异。
2 结 果
2.1 PCR反应扩增结果 以相应的引物进行的NPHS1基因和NPHS2基因PCR扩增都有较好的扩增结果,扩增片段与所设计的大小一致,无非特异性扩增条带。因此,可以进行纯化和测序。
2.2 PCR产物直接测序结果 患儿NPHS2基因的外显子及其周围的内含子均未发现突变。在患儿NPHS1基因的第8外显子发现一杂合突变G928A,遗传密码子由GAC变为AAC,该突变导致310位的天冬氨酸被天冬酰胺替代(Asp310Asn,D310N)。患儿母亲NPHS1基因的第8外显子也存在同一突变,而其父的NPHS1基因的第8外显子测序结果与野生型相同。同时检测到患儿NPHS1基因的第11内含子5'+1剪接位置的鸟嘌呤变为腺嘌呤(IVS11+1 G>A)的杂合突变(图1)。患儿父亲NPHS1基因的第11内含子也存在同一突变,而其母的NPHS1基因的第11内含子测序结果与野生型相同。在50例对照人群中未发现NPHS1的G928A(D310N)突变,也未发现IVS11+1 G>A突变。
3 讨 论
国外研究认为大部分CNS是由于编码肾小球裂孔隔膜关键蛋白的基因突变所致,较常见的是编码nephrin蛋白的NPHS1基因突变,但国内对CNS的致病基因研究不多。本研究患儿于生后3个月内出现肾病综合征,临床符合CNS的诊断。我们的检测结果证实了中国中部地区散发性CNS也存在NPHS1基因突变。近年来有报道CNS还可由NPHS2基因突变所致[3],但本研究对先证者进行的NPHS2的全部8个外显子测序结果显示正常,未见突变,且50例健康人中也未检出以上基因突变,因此,我们认为本研究中的2个NPHS1基因的杂合突变是CNS患儿的致病突变。经检索人类基因突变数据库(HGMD)和最新的文献,未见IVS11+1 G>A杂合突变的相关报道,而G928A错义突变为我国已报道的NPHS1基因突变[8,10]。
本研究发现的CNS患儿NPHS1基因2个杂合突变中,一个为剪接位点突变(IVS11+1G>A),位于11内含子5'端剪接给位区域。突变引起异常剪接的方式主要有5种形式:剪接位点外显子(突变临近的)删除、潜在的剪接位点激活、内含子滞留、新剪接位点的产生及外显子剪接增强子的破坏。证实该突变具体导致哪种异常剪接方式需要取肾组织提取mRNA来确定,目前患儿家长拒绝行肾穿刺术,暂时不能进行进一步的研究。另一个为错义突变(G928A),导致310位的天冬氨酸被天冬酰胺替代(Asp310Asn),由NPHS1基因的此位点导致的突变发生CNS已有报道[8,10]。将患儿及其父母的NPHS1突变进行比较分析显示:第11内含子剪接突变(IVS11+1G>A)来自患儿父亲,第8外显子的错义突变(G928A)来自患儿母亲。患儿父母为NPHS1基因突变的携带者。
此外,本研究还发现患儿存在5种NPHS1基因碱基变异:E117K(rs3814995)、S1105S (rs2071327)、IVS8+68A>G、IVS24+36C>T和IVS27+45C>T,经与NCBI和Ensembl的SNP数据库比较,并检索相关文献[8,11],证实这5个变异均为单核苷酸多态性(SNPs)。
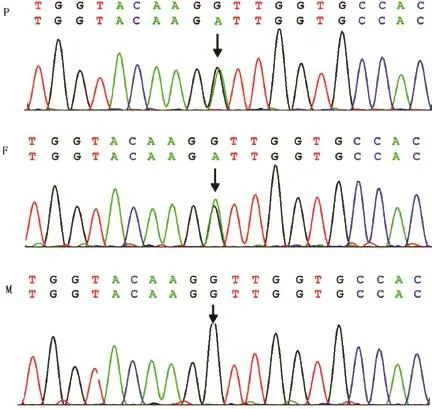
图1 新发现的CNS患儿NPHS1基因剪接位点突变测序图Fig.1 Novel splice site mutations IVS11+1G>A of the NPHS1 gene were identified by sequencing a Chinese child with congenital nephrotic syndromeChromatogram by sequencing of exon 11 of NPHS1. P. Child of CNS; F. Father of patient; M. Mother of patient; The upper:normal sequence; The lower:variation sequence; The arrows indicate mutant positions
Nephrin含有1241个氨基酸,由1个跨膜区(第1056-1241位氨基酸)和长的细胞外区域(第1-1055位氨基酸)组成,细胞外区域含有8个免疫球蛋白样基序及1个纤维连接蛋白样基序。根据NPHS1编码的分子结构,我们进一步分析了本研究所检测到的突变意义:错义突变(G928A)导致310位的天冬氨酸被天冬酰胺替代(Asp310Asn)发生在nephrin的Ig3基序(motif),而剪接位点突变(IVS11+1G>A)导致的异常剪接发生于nephrin的Ig5基序,可见2个杂合突变均影响nephrin的功能结构域(Ig样基序)。复习国内外研究结果,我们认为不同的突变部位、突变类型可能出现不同的临床表型。研究者曾报道临床表型较轻的CNS,其NPHS1基因突变位于nephrin分子的Ig1、Ig3、Ig5和Ig6基序,而临床表型较重的CNS的NPHS1突变多位于nephrin分子的Ig2、Ig4和Ig7基序[8]。造成CNS临床表现和预后不同的分子机制可能是某些NPHS1位点突变导致其编码的分子停留在内质网,不能回到足细胞膜的表面参与裂孔隔膜(SD)形成,从而出现严重的临床表现;而另一些NPHS1位点突变导致其编码的分子部分停留在内质网,尚有部分分子回到足细胞膜的表面参与SD形成,出现轻、中度的临床表现,轻、中度临床表现主要取决于回到足细胞表面参与SD形成的分子多少。而在本例患儿检测到位于NPHS1基因Ig3和Ig5基序2个杂合突变,理论上推测可能为较轻突变,我们的随访也提示,截止投稿时,该患儿已2岁10个月,肾功能尚正常,智力水平基本等同于同龄儿童,但身高及体重较同龄儿童明显降低,临床表型较轻。提示不同结构域对于nephrin分子的功能,以至发生突变后对于临床表型的影响可能不同,因此分析并确定CNS基因突变性质和位置有望指导临床客观地判断预后。
国内已有4家医院对家族性和散发性CNS进行相关基因突变分析。石岩等[8]于2005年率先在1个中国CNS家系中检测出了NPHS1基因3个杂合突变,其中1个为缺失突变(1893-1900del8),另外2个为错义突变(G928A和G2869C),证实中国CNS家系中存在NPHS1基因突变。随后,王道静等[11]在1例中国南方汉族散发性CNS患儿中检测到NPHS1基因纯合插入突变(3250insG);李国民等[12]在2例CNS患儿中检测到NPHS1基因突变:1例患儿为C3325T错义突变(R1109X,Fin-minor)和IVS26-2A>T杂合突变,1例患儿为C3325T错义突变(R1109X,Finminor)和C3478T错义杂合突变;米荣等[10]在1例CNS患儿中检测到NPHS1基因2个杂合突变(G928A 和IVS18+5G>A),合并巨细胞病毒(CMV)感染,病情较重。本研究发现了1例CNS患儿存在NPHS1基因突变,即G928A和IVS11+1G>A的复合杂合突变,IVS11+1G>A既往未见文献报道。国内尚未发现有CNS患儿存在其他基因突变的报道。NPHS1基因是否是中国CNS患儿的主要致病基因,仍需要更多的病例来证实。临床上对于CNS患儿可以先做NPHS1基因突变分析。通过复习国内相关文献,发现在3例来自中部地区的CNS患儿均检测到NPHS1基因G928A错义突变,该突变是不是中国中部地区汉族CNS的NPHS1基因的热点突变,仍有待于更多的研究来证实。
致谢 感谢南京军区福州总医院儿科陈新民、余自华教授和实验科兰风华、王水良教授在本课题设计及实验方面给予的帮助和指导!
[1]Kestilä M, Lenkkeri U, Männikkö M,et al. Positionally cloned gene for a novel glomerular protein - nephrin - is mutated in congenital nephrotic syndrome[J]. Mol Cell, 1998, 1(4):575-582.
[2]Zhang HW, Wang F, Ding J,et al. Updates on the molecular genetics of congenital nephrotic syndrome[J]. Chin J Pediatr, 2011, 49(6):425-427. [张宏文, 王芳, 丁洁, 等. 先天性肾病综合征的分子遗传学研究新进展[J]. 中华儿科杂志, 2011, 49(6):425-427.]
[3]Kari JA, Montini G, Bockenhauer D,et al. Clinico-pathological correlations of congenital and infantile nephrotic syndrome over twenty years[J]. Pediatr Nephrol, 2014, 29(11):2173-2180.
[4]McCarthy HJ, Bierzynska A, Wherlock M,et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome[J]. Clin J Am Soc Nephrol, 2013, 8(4):637-648.
[5]Bińczak-Kuleta A, Rubik J, Litwin M,et al. Retrospective mutational analysis of NPHS1, NPHS2, W T1 and LAMB2 in children with steroid-resistant focal segmental glomerulosclerosis--a single-centre experience[J]. Bosn J Basic Med Sci, 2014, 14(2):89-93.
[6]Zhang LW, Wang LP. Recent advances in the research on mechanisms underlying podocyte-specific gene mutation-related steroid-resistant nephrotic syndrome[J]. Chin J Contemp Pediatr, 2014, 16(1):99-103. [张黎雯, 王乐平. 足细胞相关突变基因致激素耐药型肾病综合征的机制研究进展[J]. 中国当代儿科杂志, 2014, 16(1):99-103.]
[7]Zhang HW, Ding J. Advances on treatment of congenital nephrotic syndrome[J]. J Appl Clin Pediatr, 2011, 26(5):373-347. [张宏文, 丁洁. 先天性肾病综合征的治疗进展[J]. 实用儿科临床杂志, 2011, 26(5):373-347.]
[8]Shi Y, Ding J, Liu JC,et al.NPHS1 mutations in a Chinese family with congenital nephrotic syndrome[J]. Chin J Pediatr, 2005, 43(11):805-809. [石岩, 丁洁, 刘景城, 等. 中国人先天性肾病综合征NPHS1基因突变[J]. 中华儿科杂志, 2005, 43(11):805-809.]
[9]Feng DN, Yang YH, Yu ZH,et al. Mutational analysis of podocyte gene in 30 Chinese children with sporadicsteroid-resistant nephritic syndrome[J]. J Appl Clin Pediatr, 2012, 27(5):327-330. [封东宁, 杨勇辉, 余自华, 等. 散发性激素耐药型肾病综合征患儿30例足细胞基因突变分析[J]. 实用儿科临床杂志, 2012, 27(5):327-330.]
[10]Mi R, Wang XY, Kang LM,et al. Mutation of NPHSl gene in an infant with congenital nephritic syndrome[J]. Beijing Med J, 2013, 35(4):253-256. [米荣, 王晓颖, 康利民, 等. 先天性肾病综合征1例报告及文献复习[J]. 北京医学, 2013, 35(4):253-256.]
[11]Wang DJ, Yu ZH, Meng DC,et al. Mutation of NPHS1 gene in a child with congenital nephritic syndrome in southern Chinese Han ethnic group[J]. J Appl Clin Pediatr, 2011, 26(5):336-338.[王道静, 余自华, 孟大川, 等. 中国南方汉族散发性先天性肾病综合征NPHS1基因突变[J]. 实用儿科临床杂志, 2011, 26(5):336-338.]
[12]Li GM, Fang XY, Xu H,et al. Two Chinese cases with congenital nephrotic syndrome caused by Fin-minor mutation of NPHSl gene and fiterature review[J]. Chin J Evid Based Pediatr, 2014, 9(1):41-44. [李国民, 方晓燕, 徐虹, 等. NPHS1基因Finminor突变导致中国先天性肾病综合征2例报道并文献复习[J]. 中国循证儿科杂志, 2014, 9(1):41-44.]
Mutation of NPHS1 gene in a Chinese child with congenital nephrotic syndrome
FU Rong, WU Qing-yan, XU Ji-xiang, GOU Meng-fan, LIU Juan, TONG Shao-feng, LIU Xue-jie, GUO Hui, HE Jun-jie
Department of Pediatrics, Puyang Oilfield General Hospital, Puyang, Henan 457001, China
ObjectiveTo analyze the mutations and characteristics of NPHS1 and NPHS2 genes in a child with congenital nephrotic syndrome (CNS).MethodsMutation analysis was made for all exons and exon/intron boundaries of NPHS1 and NPHS2 genes in a child and his parents as well as 50 unrelated adults with normal urine test results as control using PCR and direct sequencing techniques.ResultsNo mutation of NPHS2 gene was detected, while a novel splice site mutation of IVS11+1G>A within intron 11 and a missense mutation within exon 8 (c.928G>A) in NPHS1 gene were detected in the child with CNS. Urinalysis was normal in child's mother, and it was found that c.928G>A (D310N) but no IVS11+1G>A heterozygous mutation, and his father was shown to have a normal urinalysis results, and the result of gene examination was IVS11+1G>A, but without c.928G>A (D310N) heterozygous mutation. All these IVS11+1 G>A and c.928G>A (D310N) mutations were not found in the 50 unrelated controls.ConclusionsTwo heterozygote mutations IVS11+1 G>A and c.928G>A in the NPHS1 gene have been identified in a child with CNS in the central region of China. The splice site mutation of IVS11+1 G>A is an novel genetic defect of CNS. It is necessary to look for mutations in NPHS1 gene in the children with CNS.
nephrotic syndrome; genes NPHS1; mutation
R692
A
0577-7402(2015)07-0578-04
10.11855/j.issn.0577-7402.2015.07.13
2014-12-06;
2015-04-08)
(责任编辑:张小利)
付荣,副主任医师,博士研究生。主要从事小儿肾脏疾病的基础与临床研究
457001 河南濮阳 河南省濮阳市油田总医院儿科(付荣、吴清岩、徐纪香、缑梦帆、刘娟、同少峰、刘雪杰、郭辉、和俊杰)
