ZnO催化对苯二甲酸脱羧制苯的反应历程
2015-06-28蒋斌波王靖岱阳永荣
庄 岩,蒋斌波,王靖岱,阳永荣
(浙江大学 化学工程联合国家重点实验室,浙江 杭州310027)
ZnO催化对苯二甲酸脱羧制苯的反应历程
庄 岩,蒋斌波,王靖岱,阳永荣
(浙江大学 化学工程联合国家重点实验室,浙江 杭州310027)
通过比较不同金属氧化物催化对苯二甲酸脱羧制苯的活性及反应产物分布情况,优选ZnO作为脱羧催化剂。采用FT-IR、XRD和Py-GC/MS表征了催化对苯二甲酸脱羧反应前后的ZnO催化剂及对苯二甲酸锌配合物。同时还对对苯二甲酸锌配合物进行了TG-DTA及DTG分析,以探讨ZnO催化对苯二甲酸脱羧反应机理以及催化剂积炭的原因。结果表明,当反应温度为550℃、质量空速为0.48 h-1时,ZnO催化对苯二甲酸脱羧的转化率达到100%,产物中苯的质量分数达40%。ZnO催化对苯二甲酸脱羧反应分两步进行,首先对苯二甲酸化学吸附在ZnO表面形成对苯二甲酸锌配合物,然后再热分解生成苯、CO2等。对苯二甲酸锌配合物的热分解温度在410~530℃,在N2中热分解时发生的深度脱氢反应导致积炭的形成。
对苯二甲酸(TPA);氧化锌(ZnO);脱羧;苯
精对苯二甲酸(PTA)生产过程中,氧化单元排放的固体废弃物中含有大量的对苯二甲酸(TPA)等芳香酸,及少量的有色杂质和钴、锰金属催化剂等,其含量因生产工艺的不同而改变[1],导致其回收利用比较困难。通常采用焚烧、掩埋和生物降解等方法处理PTA残渣,但容易造成资源浪费和二次污染。目前,有研究者将其应用于生产增塑剂、不饱和聚酯树脂和聚酯涂料等,但其中的有色杂质和苯甲酸严重影响了产品的色泽和质量[2]。
笔者首次提出PTA残渣催化脱羧的处理方法,即将其中的芳香酸通过催化脱羧反应制备苯、甲苯等芳烃。该方法得到的产物简单,易分离精制,可以实现资源有效利用和环境保护。然而,用于该反应的多相催化剂却不多,目前仅有某些碱土金属氧化物[3]和过渡金属氧化物[4-8]可以催化芳香酸脱羧。Masuda等[6]研究了FeOOH催化对苯二甲酸脱羧,反应产物主要是二氧化碳、二苯甲酮、苯甲酸等,苯的含量较少。Yoshioka等[7-8]考察了对苯二甲酸在CaO上的催化脱羧行为,苯是主要产物,但CaO在反应后生成CaCO3,降低了催化脱羧活性,且积炭量在20%以上。现有的脱羧催化剂存在苯收率低、副产物多以及积炭量偏高的问题。
笔者考察了PTA残渣中主要组分对苯二甲酸(TPA)在不同金属氧化物催化下的脱羧行为,并采用FT-IR、XRD、Py-GC/MS等方法表征催化剂,讨论了TPA的脱羧反应机理。
1 实验部分
1.1 原料
Al(NO)3·9H2O、Zn(NO)2·6H2O、Mg(NO)2·6H2O、Ce(NO)3·6H2O、吡啶、对苯二甲酸(TPA),分析纯,国药集团化学试剂有限公司产品。
1.2 金属氧化物催化剂的制备
配制一定浓度的硝酸盐溶液,于60℃恒温下逐滴加入氨水调节溶液pH值,至出现糊状沉淀。过滤,用纯净水洗涤至中性。经110℃干燥,600℃焙烧4 h,研磨筛分,得所需粒径的金属氧化物催化剂颗粒。
1.3 对苯二甲酸锌配合物的制备
称取1.5 gTPA和4 g Zn(NO)2·6 H2O溶于15 mL吡啶中,在110℃下加热回流4 h。生成的白色沉淀物经过滤、乙醇洗涤后, 60℃真空干燥至恒重, 280℃焙烧2 h,制得白色对苯二甲酸锌配合物(TP-Zn)。
1.4 催化剂的表征
采用岛津公司XRD-6000型X射线衍射分析仪对样品进行XRD分析,管电压40 kV,管电流80 mA,CuKα射线,连续扫描,2θ扫描范围5°~90°。采用Nicolet 公司5700型红外光谱仪对样品进行FT-IR分析,KBr压片,波数范围400~4000 cm-1,分辨率4 cm-1,扫描次数32次。采用METTLER公司 TGA/SDTA型热分析仪对TP-Zn样品进行TG分析,升温速率15℃/min。采用Thermo Fisher Scientific公司CDS 5200/DSQ II型裂解色质联用仪对TP-Zn样品进行Pyrolysis-GC-MS分析。称取一定量TP-Zn样品置于微型石英裂解管中,裂解温度700℃,保持10 s。色谱柱为DB-WAX (30 m×0.25 mm×0.25 μm),载气He,分流比50, 色谱柱程序升温,从50℃(5 min)→20℃/min→260℃(15 min)。质谱分析采用EI离子源,电子能量70 eV,接口温度250℃,相对分子质量扫描范围25~500。
1.5 催化剂评价
采用固定床反应器(管直径25 mm×2.5 mm、管长1000 mm)评价催化剂催化脱羧性能,实验装置如图1所示。催化剂粒径40~80目,催化剂质量10 g,TPA总进料量4.8 g。将质量浓度为0.08 g/mL的TPA吡啶溶液,以一定的流量(MHSV=0.48 h-1)泵入固定床反应器中,在N2中于一定的温度下进行脱羧反应。生成物经冷却,气、液分离后分别收集。反应结束后,在N2吹扫下将反应器温度降至室温,取出催化剂进行表征。采用在线气相色谱仪(GC-TCD)分析气体产物组成,色谱柱为自制聚合物填充柱,柱温70℃,桥电流120 mA。采用离线气相色谱仪(GC-FID)分析液体产物组成,DB-5HT毛细管色谱柱(30 m×0.32 mm×0.10 μm),色谱柱箱程序升温,从50℃(5 min)→5℃/min→70℃(1 min)→25℃/min→250℃(3 min)。
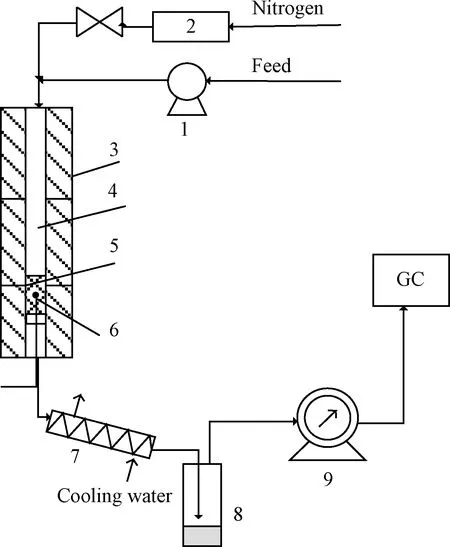
图1 管式固定床脱羧反应实验装置
2 结果与讨论
2.1 不同金属氧化物的催化脱羧性能
为了消除吡啶对TPA脱羧反应的影响,以玻璃珠为惰性物质代替催化剂进行空白实验。4种金属氧化物催化TPA脱羧反应的产物分布示于图2。由图2可以看出,在500℃时,TPA的热稳定性较高,空白实验的转化率仅为13.3%。而在ZnO、MgO、Al2O3、CeO2催化下,TPA的转化率有不同程度的提高,其中ZnO的催化脱羧活性和苯选择性最高,产物中苯的质量分数达到35%。加之价廉易得,ZnO将具有广阔的应用前景。
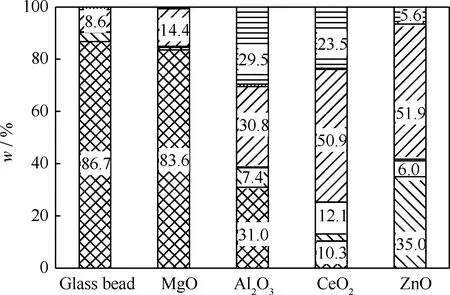
图2 500℃时4种金属氧化物催化TPA脱羧反应的产物分布
图3为TPA脱羧反应空白实验中TPA转化率和苯收率随反应温度的变化。由图3可知,在无金属氧化物催化剂时,TPA脱羧反应的TPA转化率和苯收率都随着反应温度的升高而不断增加。在TPA热分解过程中,吡啶中的N原子可能与羧酸中的羰基结合形成中间过渡态结构[9-10],从而起到催化脱羧作用。因此,TPA的热分解可认为是温度和吡啶共同作用的结果。
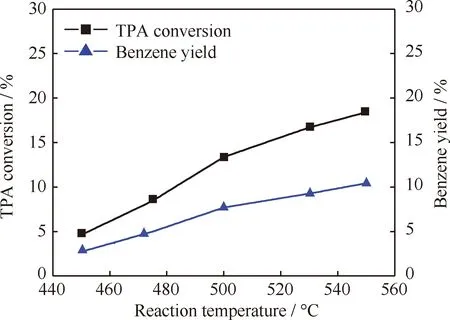
图3 TPA脱羧反应空白实验中反应温度对TPA转化率和苯产率的影响
2.2 反应温度对ZnO催化TPA脱羧反应的影响
理论上TPA在完全转化时会生成质量分数为47%的苯和53%的CO2。图4为反应温度对ZnO催化TPA脱羧反应产物分布的影响。由图4可知,随着反应温度的升高,TPA脱羧反应的转化率不断增加,产物中苯的质量分数也逐渐增加,由450℃的19.34%增至550℃的41.69%;CO2的质量分数也呈增加的趋势,由450℃的28.72%增至550℃的52.36%;而残余TPA质量分数逐渐降低,由450℃的25.74%降至550℃的1.85%。反应产物中均含有苯甲酸,其含量随反应温度的升高先增加后降低。因此,在550℃下,ZnO催化TPA脱羧反应的活性最高,且副产物含量少。
随着反应温度的升高,TPA脱羧产物中苯、苯甲酸含量先同时增加,随后苯甲酸含量逐渐降低,而苯含量却继续增加。由此可以说明,苯甲酸是TPA脱羧反应的中间生成物,它可以进一步脱羧生成苯。可以认为,TPA的催化脱羧反应同时存在串联反应和一步反应。串联反应即TPA首先脱去1个羧基生成苯甲酸,然后苯甲酸继续催化脱羧生成苯和CO2等产物;一步反应即TPA直接催化脱羧生成苯和CO2等产物。

图4 反应温度对ZnO催化TPA脱羧反应产物分布的影响
2.3 对苯二甲酸催化脱羧反应机理
对脱羧反应机理的深入研究,有助于解决催化剂的积炭问题和减少副反应的发生,并对催化剂配方的优选提供指导。一般认为,金属氧化物催化乙酸脱羰基制丙酮[11-12]反应过程中,乙酸首先吸附在金属氧化物表面生成羧酸盐中间物,然后经脱水、烯酮化等历程生成丙酮。金属氧化物催化TPA脱羧反应过程中也有可能生成羧酸盐。因此,笔者采用FT-IR、XRD、Py-GC/MS表征了催化TPA脱羧反应前后的ZnO催化剂和对苯二甲酸锌配合物TP-Zn。
图5为TPA、TP-Zn和450℃催化TPA脱羧反应后ZnO的FT-IR谱。由图5可看出,TPA在1678 cm-1处有C=O振动引起的吸收峰和在2500~3000 cm-1范围内由O—H振动引起的较宽的吸收峰。TP-Zn在以上位置无明显的吸收峰,而在1591 cm-1和1386 cm-1处出现了2个新的吸收峰[13]。羧酸盐的羧基—COO-具有多电子π键体系,2个C—O振动频率相近,有强烈的振动偶合作用,结果导致1678 cm-1处C=O 的振动峰消失,在1591 cm-1和1386 cm-1处出现2个新的吸收峰[13]。以上结果表明,—COOH上的氢解离后以羧酸根的形式存在。TP-Zn在3200~3600 cm-1范围内较宽的吸收峰[14]则是由于其表面存在吸附水而引起。450℃催化TPA脱羧反应后的ZnO的红外吸收峰位置与TP-Zn的特征峰位置相同,表明TPA可能在ZnO表面被化学吸附生成了TP-Zn。
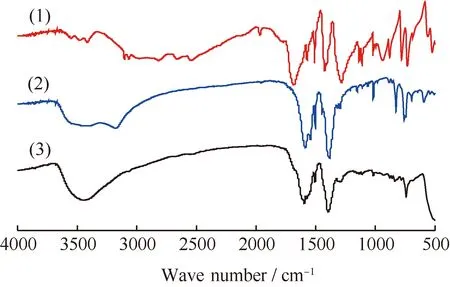
图5 TPA、TP-Zn和450℃催化TPA脱羧反应后ZnO的FT-IR谱
图6为TP-Zn和450℃催化TPA脱羧反应前后ZnO的XRD谱。由图6可以看出,450℃催化TPA脱羧反应后的ZnO与催化反应前ZnO相比较,在2θ为10.1°、16.4°、18.0°、23.5°、30.1°处出现了新的衍射峰。这些衍射峰的位置与TP-Zn的特征衍射峰相同。此结果进一步表明,TPA在脱羧反应过程中在ZnO表面生成了TP-Zn。图7为不同温度催化TPA脱羧反应后ZnO与新鲜ZnO的XRD谱。对比图6和图7可以看出,500℃反应后ZnO的XRD谱中并没有TP-Zn的特征峰,其衍射峰的位置与纯ZnO一致,表明在500℃反应时ZnO表面的TP-Zn分解完全,且没有新的晶型出现。
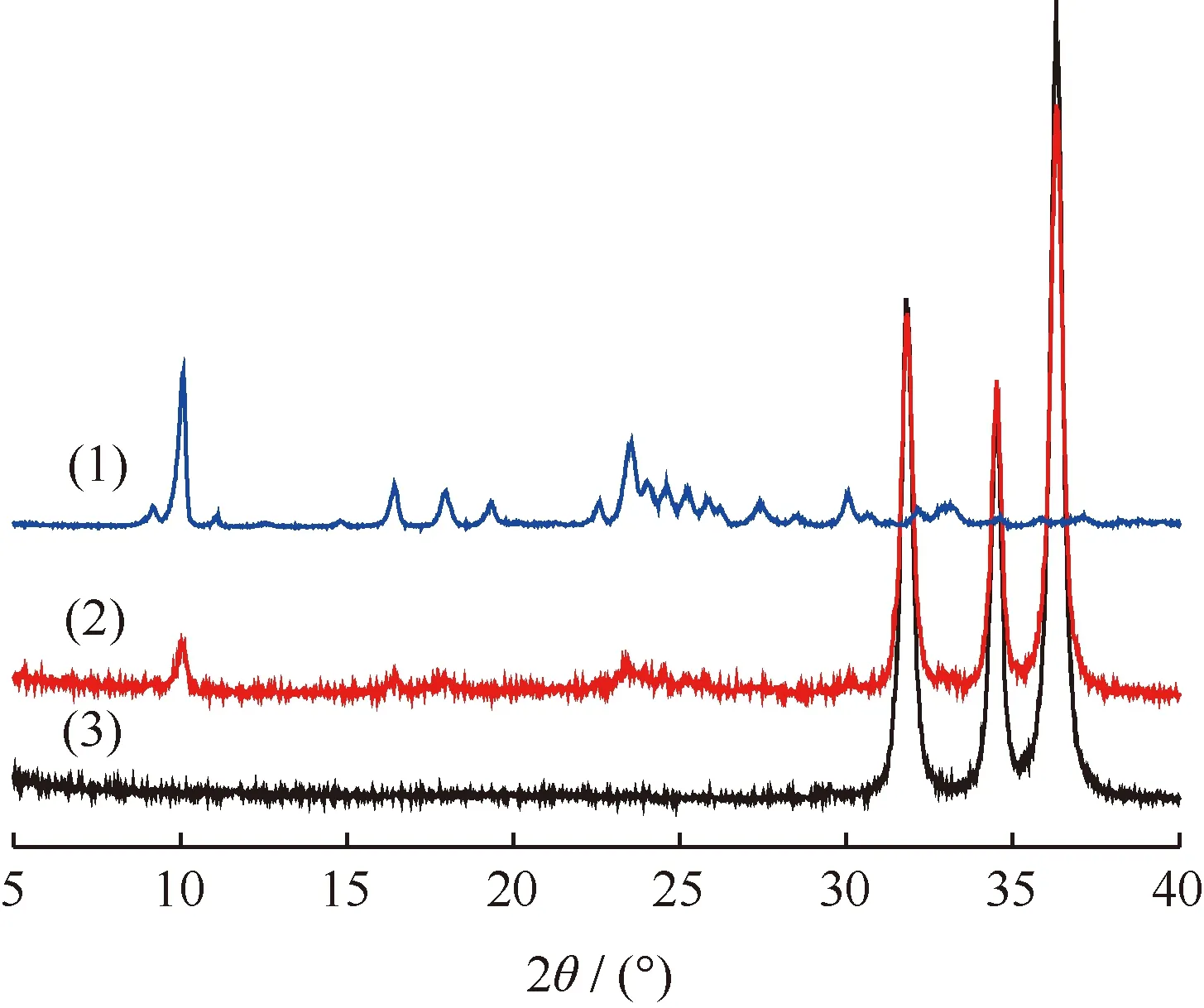
图6 TP-Zn和450℃催化TPA脱羧反应前后ZnO的XRD谱
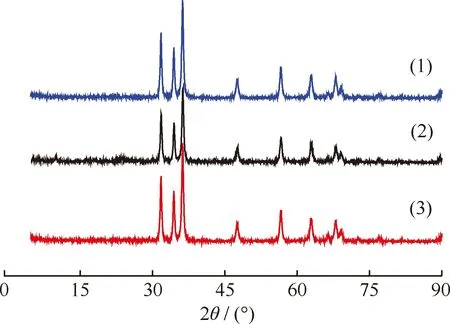
图7 不同温度催化TPA脱羧反应后ZnO与新鲜ZnO的XRD谱
图8为TP-Zn的Pyrolysis-GC-MS总离子流图。利用标准质谱库检索的信息对主要的13个峰所对应的裂解产物进行定性分析,所得定性、定量结果列于表1。从图8和表1可见,TP-Zn裂解产物中苯的含量占绝对优势,其次是吡啶、二苯甲酮、联苯等。吡啶是TP-Zn样品制备过程中被吸附的溶剂。ZnO催化TPA脱羧得到的产物中主要是苯和CO2,此外还含有少量的二苯甲酮、联苯等,与TP-Zn裂解产物相同。因此,TP-Zn是对苯二甲酸在ZnO催化下脱羧反应过程中的中间生成物。
由FT-IR、XRD和Pyrolysis-GC-MS分析结果可以推出TPA的脱羧反应机理,如图9所示。即TPA首先化学吸附在ZnO表面生成TP-Zn,然后TP-Zn在高温下热分解生成苯、CO2等产物。
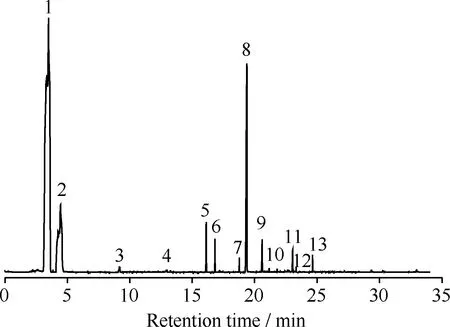
图8 TP-Zn的Pyrolysis-GC-MS总离子流色谱
表1 TP-Zn的Pyrolysis-GC-MS分析结果
Table 1 Analysis results from Pyrolysis-GC-MS of TP-Zn
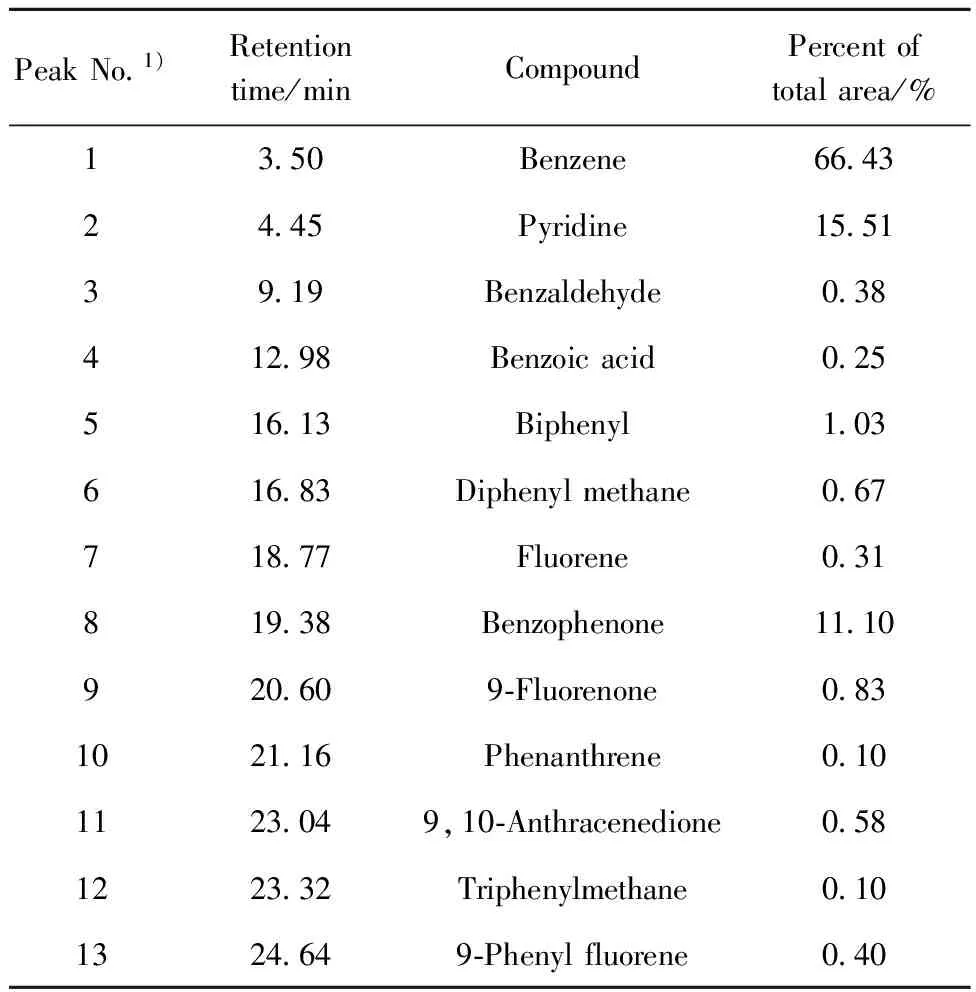
PeakNo 1)Retentiontime/minCompoundPercentoftotalarea/%13 50Benzene66 4324 45Pyridine15 5139 19Benzaldehyde0 38412 98Benzoicacid0 25516 13Biphenyl1 03616 83Diphenylmethane0 67718 77Fluorene0 31819 38Benzophenone11 10920 609⁃Fluorenone0 831021 16Phenanthrene0 101123 049,10⁃Anthracenedione0 581223 32Triphenylmethane0 101324 649⁃Phenylfluorene0 40
1) Shown in Fig.8
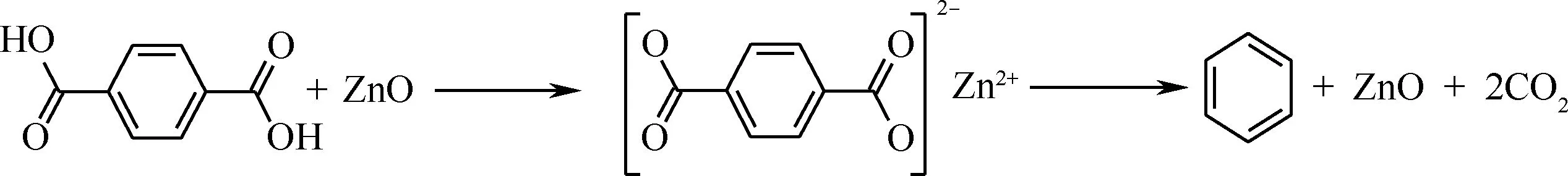
图9 TPA在ZnO催化下的脱羧反应机理
2.4 ZnO催化TPA脱羧反应积炭形成的原因
TP-Zn的热分解是TPA 脱羧反应过程中的重要环节,其热分解行为与积炭的形成密切相关。图10为TP-Zn在空气和N2中的TG-DTA曲线,图11为相应的DTG曲线。由图10可发现,TP-Zn在空气和N2中热分解的趋势有所区别。在空气中,TG曲线有3个失重阶段,DTA曲线存在2个较弱的吸热峰和1个强放热峰。DTA曲线在203.92℃处的弱吸热峰对应的失重,归属于样品骨架中束缚水及溶剂的脱除;526.03℃处的强放热峰对应的失重则是有机配体在空气中燃烧反应的结果。在N2中,TG曲线有4个失重阶段,DTA曲线存在3个较弱的吸热峰和2个弱放热峰。DTA曲线在195.3℃处的弱吸热峰对应的失重,归属于样品骨架中束缚水及溶剂吡啶的脱除;530.94℃处的弱放热峰对应的失重则是TP-Zn热分解反应的结果。TP-Zn在空气和N2中的失重量不同。TP-Zn与空气中的O2反应,最终生成ZnO,530℃后质量达到稳定,失重量67.3%;程序升温至530℃时,TP-Zn在N2中的失重量59.9%,明显低于前者。TP-Zn裂解产物中含有少量的芴、9,10-蒽醌、菲等(见表1),可见,在N2中,TP-Zn同时进行了热分解反应和深度脱氢反应,最终生成C/ZnO复合产物。TP-Zn深度脱氢反应生成的焦炭与ZnO可能发生氧化还原反应,导致TP-Zn在N2中程序升温至1000℃时的失重量(68.7%)高于其在空气中的失重量(67.4%)。
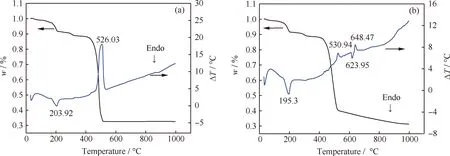
图10 TP-Zn在空气和N2中的TG-DTA曲线
由图11可以看出,TP-Zn在300℃时,溶剂和水已基本完全脱除;TP-Zn在空气和N2中的热解温度均在410~530℃之间;随着温度的升高,TP-Zn的热分解速率加快,且在500℃左右失重速率达到最大值。以ZnO为催化剂在较低温度,如450℃下催化TPA脱羧反应时, 其中间产物TP-Zn未分解完全,因此而导致ZnO催化剂的表观积炭量偏高(见图4)。
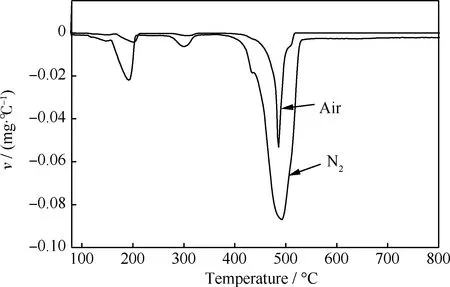
图11 TP-Zn在空气和N2中的DTG曲线
3 结 论
(1) ZnO具有较好的催化TPA脱羧制苯性能,脱羧反应主要产物是苯和CO2。在反应温度550℃、常压和质量空速0.48 h-1条件下,TPA转化率高达100%,产物中苯的质量分数可达40%,积炭量约为2%。
(2) ZnO催化TPA的脱羧反应历程分为两步,首先TPA在ZnO表面被化学吸附生成对苯二甲酸锌配合物,然后继续热分解生成苯、二氧化碳等。
(3) 对苯二甲酸锌配合物在空气和N2中的热分解温度均在410~530℃之间,其DTG峰对应的温度为500℃。其在N2中热分解时,还能发生深度脱氢反应,导致催化剂积炭。
[1] DIESSEL K, MODIC R, STRUSS F. Recovery and reuse of heavy-metal oxidation catalyst from the Witten DMT process:US, 4410449[P].1983-10-18.
[2] ROFFIA P, CALINI P, MOTTA L, et al. Byproduct identification in the terephthalic acid production process and possible mechanisms of their formation[J]. Industrial & Engineering Chemistry Product Research and Development, 1984, 23(4):629-634.
[3] ZHANG A, MA Q,WANG K, et al. Naphthenic acid removal from crude oil through catalytic decarboxylation on magnesium oxide[J]. Applied Catalysis A:General, 2006, 303(1):103-109.
[4] DE LANGE M W, VAN OMMEN J G, LEFFERTS L. Deoxygenation of benzoic acid on metal oxides 1 The selective pathway to benzaldehyde[J]. Applied Catalysis A:General, 2001, 220(1-2):41-49.
[5] DE LANGE M W, VAN OMMEN J G, LEFFERTS L. Deoxygenation of benzoic acid on metal oxides 2 Formation of byproducts[J]. Applied Catalysis A:General, 2002, 231(1):17-26.
[6] MASUDA T, MIWA Y, HASHIMOTO K, et al. Recovery of oil from waste poly(ethylene terephthalate) without producing any sublimate materials[J]. Polymer Degradation and Stability, 1998, 61(2):217-224.
[7] KUMAGAI S, GRAUSE G, KAMEDA T, et al. Decomposition of gaseous terephthalic acid in the presence of CaO[J]. Industrial & Engineering Chemistry Research, 2011, 50(4):1831-1836.
[8] GRAUSE G, HANDA T, KAMEDA T, et al. Effect of temperature management on the hydrolytic degradation of PET in a calcium oxide filled tube reactor[J]. Chemical Engineering Journal, 2011, 166(2):523-528.
[9] ARTOK L, SCHOBERT H H. Reaction of carboxylic acids under coal liquefaction conditions 1 Under nitrogen atmosphere[J]. Journal of Analytical and Applied Pyrolysis, 2000, 54(1):215-233.
[10] CLARK L W. The mechanism of the decomposition of trichloroacetic acid in aromatic amines[J]. The Journal of Physical Chemistry, 1959, 63(1): 99-101.
[11] RAJADURAI S. Pathways for carboxylic acid decomposition on transition metal oxides[J]. Catalysis Reviews, 1994, 36(3): 385-403.
[12] VOHS J M, BARTEAU M A. Reaction pathways and intermediates in the decomposition of acetic and propionic acids on the polar surfaces of zinc oxide[J]. Surface Science, 1988, 201(3):481-502.
[14] BRZYSKA W, WOODKIEWICZ W. Thermal decomposition of copper (II) benzenedicarboxylates[J]. Journal of Thermal Analysis and Calorimetry, 1988, 34(5):1207-1215.
Catalytic Decarboxylation Mechanism of Terephthalic Acid to Benzene Over ZnO Catalyst
ZHUANG Yan, JIANG Binbo, WANG Jingdai, YANG Yongrong
(StateKeyLaboratoryofChemicalEngineering,ZhejiangUniversity,Hangzhou310027,China)
Several metal oxides catalysts were prepared for catalytic decarboxylation of terephthalic acid(TPA), and then ZnO was selected after comparison of their catalytic activities and reaction products distribution of TPA decarboxylation. FT-IR, XRD and Py-GC/MS were used to characterize the ZnO samples before and after catalytic decarboxylation and zinc terephthalate (TP-Zn), which was also analyzed by TG-DTA and DTG, to study the mechanism of the decarboxylation and coke formation. The results showed that conversion of TPA decarboxylation over ZnO was about 100% and the content of benzene in products was over 40% at 550℃ and MHSV of 0.48 h-1. TP-Zn intermediate was identified on ZnO after its catalyzing decarboxylation. The TPA decarboxylation over ZnO involved two steps, in which the first step was chemical adsorption of TPA on ZnO to form TP-Zn and the second step was thermal decomposition of TP-Zn to produce benzene and CO2. The decomposition temperature of TP-Zn was between 410℃ and 530℃ in N2atmosphere. The decomposition companied with deep dehydrogenation of TP-Zn resulted in carbon deposition on ZnO surface.
terephthalic acid(TPA);zinc oxide(ZnO);decarboxylation;benzene
2013-12-17
国家自然科学基金项目(21176208)和国家高技术研究发展计划项目(2012AA030304)资助
庄岩,男,硕士研究生,从事工业催化研究;E-mail:zhuangzi7295@126.com
蒋斌波,男,副教授,博士,从事多相流反应工程、多相流检测与信息处理研究;Tel:0571-87952254; E-mail:jiangbb@zju.edu.cn
1001-8719(2015)03-0698-07
TQ032
A
10.3969/j.issn.1001-8719.2015.03.013
