用于制备生物柴油的固体酸催化剂的研究进展
2015-06-28刘亚录朱运培袁忠勇
刘亚录,朱运培,袁忠勇
(南开大学 先进能源材料化学教育部重点实验室 天津化学化工协同创新中心,天津 300071)
用于制备生物柴油的固体酸催化剂的研究进展
刘亚录,朱运培,袁忠勇
(南开大学 先进能源材料化学教育部重点实验室 天津化学化工协同创新中心,天津 300071)
固体酸催化剂由于其易于分离回收、对生产设备无腐蚀性、无毒、对环境友好等优势而应用于生物柴油的制备。综述了当前应用于生物柴油制备的固体酸催化剂的研究进展,包括沸石、杂多酸、强酸性离子交换树脂、硫酸化金属氧化物固体超强酸、金属磷酸盐和有机膦酸盐杂化材料,以及新型的碳基固体酸催化剂,讨论了它们在生物柴油制备等酸催化反应中存在的问题,并展望其发展方向。
生物柴油;固体酸;酸催化
随着全球经济的高速发展,化石燃料的大量燃烧给环境和人们的生活造成了严重的影响,加之其资源有限,让人们越来越意识到环境污染和能源危机的严重性,发展可持续的绿色能源受到越来越多的关注[1-3]。生物柴油作为一种绿色、可再生的资源,具有优良的燃烧性能、润滑性能、安全性能等优点,有望取代传统的化石燃料应用于人类的生产生活中[4]。生物柴油主要由游离的脂肪酸或动植物油脂与短链的醇在酸或碱催化剂催化下,发生酯化反应或酯交换反应来制备,即生物柴油为脂肪酸甲酯或乙酯等的混合物。
可用于生产生物柴油的原料很广,如油菜籽、大豆油等食用油和不可食用的原料油如废弃的食用油、麻风树籽油等。相比较而言,废弃的食用油、麻风树籽油等由于其低成本和高产率更适用于生物柴油的生产[5]。传统生物柴油生产工艺大多采用均相强碱催化剂[6-7],在比较温和的条件下就能得到较好的催化效果。但碱催化剂对原料油中的含水量及游离的脂肪酸比较敏感,若原料油中脂肪酸含量较高,碱催化剂容易发生皂化反应,在一定程度上限制了强碱性催化剂在制备生物柴油中的应用[8]。为了克服碱催化剂的劣势,在采用较高脂肪酸含量的原料油时,往往先用酸催化剂对原料油进行酯化处理,降低原料油中游离脂肪酸的含量,之后再采用碱催化剂来制备生物柴油[9]。此外,酸催化剂能同时催化酯化反应和酯交换反应,且可以避免皂化反应的发生,故可以直接用于低成本的高酸值原料油制备生物柴油。均相酸催化剂(如硫酸、盐酸等)由于其强酸性而具有较高的酸催化活性,但是催化剂难以回收重复利用,对设备有严重的腐蚀性,且在水洗过程中要产生大量的废水,对环境造成严重污染,不能广泛应用于工业生产领域[10]。固体酸催化剂由于其易于分离回收、对生产设备无腐蚀性、无毒、对环境友好等优势,近些年来已引起研究者们的广泛关注[11-12]。已有若干关于固体酸催化剂在生物柴油制备中应用的综述[5,13-15],但大多局限于传统的固体酸催化剂如沸石、离子交换树脂、杂多酸等。笔者在总结传统固体酸催化剂在生物柴油制备中的最新研究进展的同时,详细综述了金属膦酸盐、碳基固体酸等新型固体酸在生物柴油制备中的研究进展,分析了各类型固体酸催化剂的特性和存在问题,并展望了固体酸催化剂在生物柴油制备等酸催化反应中的发展方向。
1 固体酸在生物柴油制备中的应用
1.1 沸石及介孔沸石催化剂
沸石是通过氧原子桥联起来的晶体硅铝酸盐,它们可具有不同的晶体结构、尺寸、骨架硅/铝比和质子交换能力[15]。但是,传统的沸石催化剂在生物柴油的制备过程中,由于其孔道尺寸较小,不利于长链脂肪酸及甘油三酯等大分子的吸附和扩散,往往得到较低的转化率[16]。如以H-ZSM-5、Y和Beta分子筛作为催化剂时,在醇/酸摩尔比为1、催化剂用量为3%(质量分数)、反应温度为130℃、反应时间为2 h的条件下,月桂酸酯化的转化率为40%左右[17]。这可能是由于沸石较小的孔道尺寸阻碍了较大的反应物分子的扩散,催化反应主要发生在催化剂的外表面,从而使其催化效果不太理想。因此,为了进一步提高沸石对制备生物柴油的催化效率,介孔沸石受到了研究者们的关注。在沸石合成过程中,使用硬模板或软模板(超分子模板)作为表面活性剂,或者对合成的沸石进行后处理,可以在沸石之中产生介孔,从而有利于大分子的吸附和扩散。
采用沸石晶体脱铝的方法能够在沸石晶体内形成空穴缺陷,从而产生介孔性质的二次孔,有利于涉及大分子的反应[18]。采用脱铝方法改变沸石骨架组成而制备得到的超稳Y型沸石(简称USY)是一种介孔沸石[19]。将铵型超稳Y沸石NH4USY经过焙烧热处理后得到氢型HUSY沸石,N2吸附-脱附测试表明其平均孔径为2.28 nm。这些在超稳化处理过程中产生的介孔减少了反应物分子的迁移阻力,使其容易到达活性位点。将HUSY沸石作为催化剂,应用于大豆油和乙醇的酯交换反应,在反应温度200℃、压力2×106Pa、醇/油摩尔比30的条件下反应24 h,脂肪酸酯的产率为99.7%[20];采用经溶液浸渍法得到的Ce/HUSY为催化剂,在同样条件下脂肪酸酯产率为99.8%。HUSY与Ce/HUSY的总酸位点分别为0.88和0.78 mmol/g。在酯交换反应初始的4 h内,HUSY沸石催化所得转化率更高,这可能是由于HUSY具有更多的酸位点,在反应中体现出更高的反应速率;在反应6~8 h,HUSY与Ce/HUSY催化酯交换反应得到相近的转化率;在反应进行12 h后,Ce/HUSY催化酯交换反应的转化率超过HUSY沸石的催化效果。可能是由于Ce物种在HUSY沸石表面均匀分布,有利于沸石结构的稳定,进而使酸活性位点得到稳定,这与Ce/HUSY沸石具有更好的重复使用性能一致。在重复使用3次后,HUSY沸石催化所得转化率由99.7%降至96.4%,而Ce/HUSY沸石所得转化率仍为99.5%。在重复使用后,HUSY和Ce/HUSY 2种沸石的酸位点都有所减少,但后者的活性位点的保持更多些,可能是Ce物种均匀分布在沸石表面所致。Zhang等[21]以有机硅烷化的二氧化硅作为硅源,利用Bond-blocking 原则,制备出介孔Beta沸石,之后经与NH4NO3溶液的离子交换及焙烧处理后,得到H型介孔Beta沸石,将其应用于蔬菜油与甲醇的酯交换反应中。在反应温度423 K、醇/油摩尔比15、催化剂的用量为蔬菜油质量的2%的条件下,采用传统的微孔Beta沸石催化剂时,在反应3 h、6 h和27 h后,甘油三油酸酯的转化率分别为6.44%、11.0%和16.6%;采用介孔HBeta沸石催化剂时,甘油三油酸酯的转化率分别提升至15.5%、71.3%和100%。由于沸石中介孔的引入,有利于反应物分子与酸活性位点的接触,同时也在一定程度上增加了路易斯酸位点的含量,从而表现出高于传统沸石催化剂的催化活性。
由此可见,在生物柴油制备中采用介孔沸石催化剂,在一定程度上促进了酯交换反应,但仍需较高的反应温度和反应压力。因此,如何进一步改善催化剂的结构性质,提高其在更加温和条件下的酸催化效率,仍是研究者们需要进一步关注的问题。
1.2 杂多酸催化剂
杂多酸(HPA)是由杂原子(如P、Si、Fe等)和多原子(如W、V、Nb、Ta等)通过氧原子配位桥按一定的结构组成具有八面体基本结构的多原子氧酸盐,具有较强的B酸性、无毒、对环境污染少等优点,被广泛应用于酯化[22]、酯交换[23]等酸催化反应中。Keggin结构的杂多酸化合物是最常用的杂多酸型催化剂,也是酸催化活性最高的杂多酸。在20多种杂多酸(盐)中,12-磷钨酸的催化活性最高[24]。Fernandes等[25]以磷钨酸H3PW12O40(HPW)为催化剂催化油酸与甲醇的酯化反应,在反应温度328 K、油酸0.66 mmol、甲醇6 mL、催化剂用量0.0059 mmol的条件下反应6 h,油酸甲酯的产率为99%,且重复使用6次后,油酸甲酯的产率仍在97%~98%范围,显示出较好的重复使用性能。
由于杂多酸容易在反应中形成液相体系而不便于回收,将杂多酸负载到不同的无机或有机-无机多孔材料载体上制备成固相催化剂[26-29],可使催化剂回收重复使用,同时也在一定程度上改善了其结构性质,进一步提高催化活性。Oliveira等[30]以ZrO2为无机载体,浸渍法负载不同量的H3PW12O40,所得H3PW12O40/ZrO2催化剂用于油酸与乙醇的酯化反应中;H3PW12O40负载量为20%时,在反应温度100℃、油酸/乙醇摩尔比1/6,催化剂用量10%的条件下反应4 h,油酸的转化率可达88%。反应后的H3PW12O40/ZrO2催化剂用乙醇清洗及100℃烘干后重复使用,考察其重复使用情况。结果表明,在循环使用4次后催化活性明显下降,油酸的转化率降至20%左右;而当催化剂用正己烷洗涤并在300℃下焙烧4 h后再重复使用时,在循环使用4次后,油酸的转化率保持在40%以上。这可能是由于杂多酸较易溶于极性溶剂而流失,当以非极性溶剂正己烷作为清洗剂时,催化剂稳定性得到一定程度的保持,其重复使用性能得到改善。这也表明,在重复使用中催化剂的处理方法值得进一步关注和研究,以提高固体酸催化剂的稳定性。
使用具有较高比表面积的材料作为载体时,载体的结构和性质也影响H3PW12O40的负载及其与载体的相互作用。Brahmkhatri等[31]以具有较高比表面积的介孔结构MCM-41为载体,浸渍法制备了不同负载量(10%、20%、30%和40%)的H3PW12O40/MCM-41催化剂,并以月桂酸与1-丁醇的酯化反应考察其催化活性。在反应温度90℃、脂肪酸/醇摩尔比1/2、催化剂用量0.2 g、反应时间3 h的条件下,30% H3PW12O40/MCM-41为催化剂,月桂酸酯的产率可达95%,且催化剂重复使用4次后,月桂酸酯的产率仍无明显变化。H3PW12O40高度分散在MCM-41的六方孔道中,且在MCM-41与H3PW12O40分子之间可能存在一定的化学相互作用,从而加强了该负载型催化剂的稳定性和重复使用性能。
采用表面后修饰的方法能够有效地制备负载型杂多酸HPA催化剂。然而这种方法也存在一定的缺陷,如,对HPA的实际负载量难以控制;不可避免的HPA的流失;在进行后修饰的同时,往往使材料的孔径减小,抑制了反应物和产物分子在孔道中的传输和迁移,从而在一定程度上影响了催化活性[32]。相比较而言,采用一步溶胶-凝胶共缩合的方法能有效地克服上述一些问题。Su等[33]采用一步溶胶-凝胶共聚缩合的方法制备出一系列杂多酸(H3PW12O40)和疏水烷基基团(苯基末端结合的有机硅烷试剂以及乙基或苯基桥联的有机硅烷试剂)修饰的ZrO2基有机-无机杂化材料H3PW12O40/ZrO2-Si(Ph)、H3PW12O40/ZrO2-Si(Et)Si和H3PW12O40/ZrO2-Si(Ph)Si,并将其作为固体酸催化剂应用于乙酰丙酸的酯化反应中。使用不同的有机硅烷前驱体(BTESB、BTMSE或PhTMS等),以及调变有机硅前驱体与金属锆源的初始摩尔比,所制得的杂合催化剂具有不同的结构有序性和孔道结构。如对于苯基桥联的杂化材料,当Si/Zr的初始摩尔比为1.0和1.5时,得到的杂合材料具有有序的二维六方介孔结构;当Si/Zr的初始摩尔比小于1.0时,得到具有无序三维蠕虫孔状的介孔结构的杂合材料。当Si/Zr的初始摩尔比相同时,不同有机硅烷试剂前驱体的结构也会直接影响所得杂合材料的孔道结构。如当Si/Zr的初始摩尔比为1.0,使用乙基桥联的或苯基终端结合的有机硅烷试剂取代苯基桥联的有机硅作为前驱体时,所得到的杂合催化剂呈现出三维连通的多孔孔道结构。这可能是由于苯基具有刚性结构,从而在除去材料中的结构导向剂P123后,仍能保持有序的介孔结构;相比较而言,乙基基团具有较大的灵活性,从而在除去P123后,导致材料的结构变形,得到无序的介孔结构;当以苯基终端结合的有机硅烷试剂作为前驱体时,苯基主要分布在所得材料的孔道结构中而不是材料的孔墙中,在P123被除去后,材料由于缺乏足够的结构刚性,而无法得到有序的介孔结构(见图1)。将不同初始Si/Zr摩尔比合成得到的H3PW12O40/ZrO2-Si(Ph)Si以及无烷基修饰的H3PW12O40/ZrO2用于催化乙酰丙酸与甲醇的酯化反应,比较它们的催化活性。在反应温度65℃、乙酰丙酸/甲醇摩尔比1/7、催化剂用量2%的条件下反应3 h后,乙酰丙酸甲酯的产率随着H3PW12O40/ZrO2-Si(Ph)Si催化剂的Si/Zr摩尔比的增加而提高,直至平衡;当催化剂的Si/Zr摩尔比由0.4增加至1.0时,催化所得乙酰丙酸甲酯的产率由76.4%增加到99.9%,Si/Zr摩尔比继续增加至1.5时,其产率不再明显变化。在相同反应条件下,H3PW12O40/ZrO2呈现出最低的催化活性,乙酰丙酸甲酯的产率只有40%左右。H3PW12O40/ZrO2-Si(Ph)Si杂合材料具有较高的催化活性主要是由于其具有较强的质子酸和路易斯酸位(H3PW12O40/ZrO2),以及较好的孔性结构和加强的疏水性表面。在相同的反应条件下,H3PW12O40/ZrO2-Si(Ph)Si-1.0比H3PW12O40/ZrO2-Si(Et)Si-1.0呈现出更高的催化活性,可能是由于苯基基团的疏水性强于乙基基团。此外,这种新型的有机-无机杂合催化剂在重复使用3次后,催化活性损失低于20%。其较好的催化稳定性主要取决于Keggin单元与ZrO2等载体的强共价键作用以及材料表面疏水性的增强。由此可见,杂合材料中疏水性烷基基团的引入,对其酸催化活性和稳定性有着重要的影响。但是由于金属前驱体与有机硅前驱体的水解速率不同,以及杂多酸易分解等原因,使得该催化剂的制备过程比较复杂。
以磁性纳米粒子作为载体,采用一步法可以合成得到含有杂多酸和疏水性基团的新型核壳结构的固体酸催化剂[34],其中,磁性纳米粒子四氧化三铁和二氧化硅为壳层,杂多酸与有机基团为核心。由于酸位点与疏水基团的同时存在,这种新型的固体酸催化剂具有较高的催化酯化反应活性和耐水性。在反应温度65℃、甲醇/棕榈酸摩尔比6、催化剂用量8.2%的条件下反应2 h,棕榈酸的转化率达到90.4%,TOF值为4.95×102h-1。值得注意的是,由于磁性粒子的引入,在磁场作用下,催化剂很容易从反应混合物中分离;经过5次循环使用后,催化活性仍无明显损失,表明这种新型的固体酸催化剂具有较高的稳定性和重复使用性能。在催化剂中引入磁性材料可以有力地促进材料的回收,减少催化剂的损失,这种新的设计思路对设计新型的催化剂材料具有重要的指导意义。
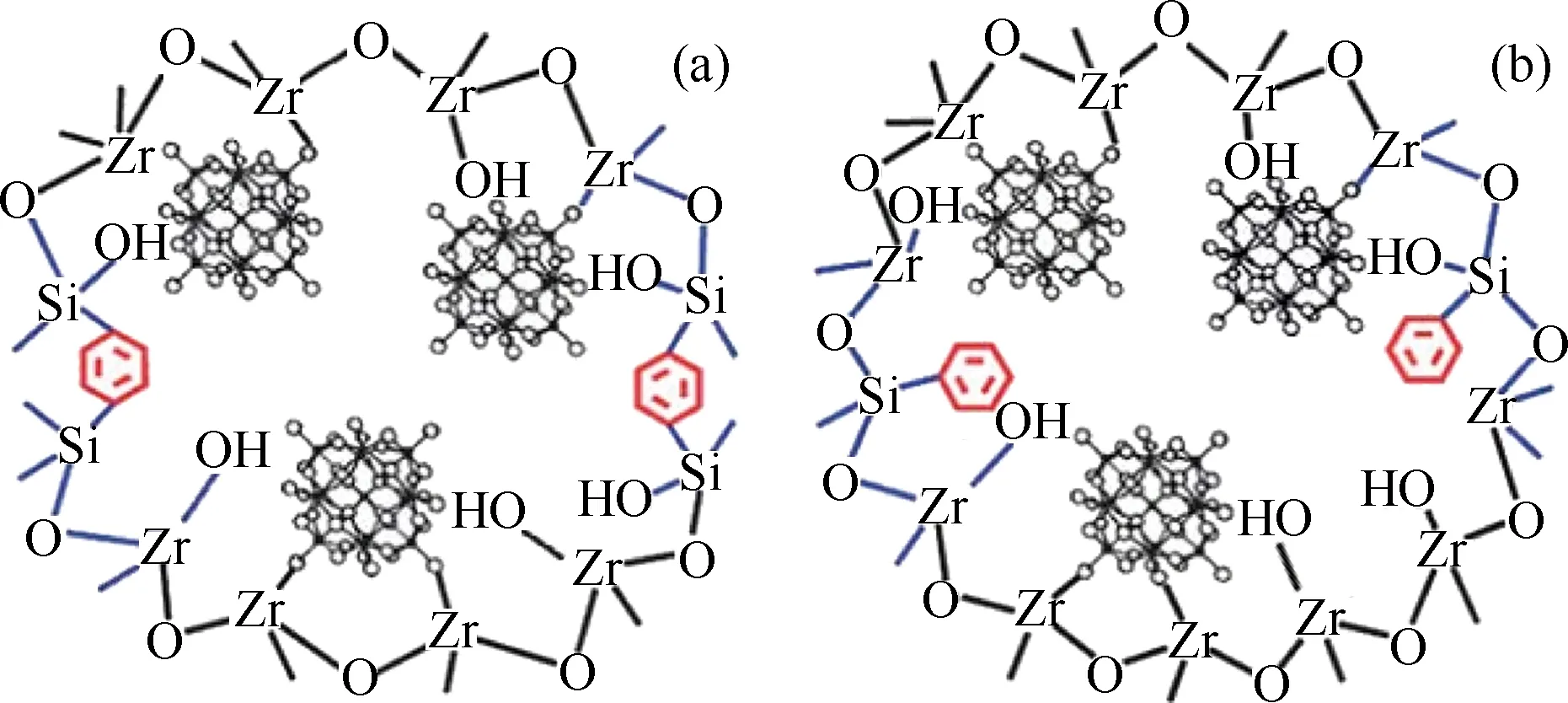
图1 H3PW12O40/ZrO2-Si(Ph)Si和H3PW12O40/ZrO2-Si(Ph) 杂合催化剂的孔墙组成示意图
1.3 强酸性离子交换树脂催化剂
在强酸性离子交换树脂中,大孔树脂如Amberlyst-15、Amberlyst-16、Amberlyst-35、Amberlyst-36、Amberlyst-131等[35-36]和全氟磺酸树脂如Nafion SAC-13、NR50等[37-38]常被作为固体酸催化剂,应用于酯化反应和酯交换反应中。如,在反应温度150℃、月桂酸/2-乙基己醇摩尔比1、催化剂用量3%、反应时间2 h的条件下,以Amberlyst-15和NR50作为催化剂,月桂酸的转化率分别可达到98%和78%[17]。这2种树脂催化剂在反应的初始阶段都体现出较高的催化活性,但是,在反应2 h后Amberlyst 15就失活,反应4.5 h后NR50也失活。这可能是由于反应温度比较高的缘故。Ilgen[39]考察了Amberlyst 46树脂催化剂在油酸和甲醇的酯化反应中的酸催化活性,在反应温度100℃、甲醇/油酸摩尔比3、催化剂用量15%的条件下反应2 h,油酸的转化率可达98.6%,且Amberlyst 46在重复使用10次后,催化活性仍没有明显变化,具有较好的稳定性。在反应温度65℃、甲醇/废弃食用油WCO摩尔比15、Amberlyst 15催化剂用量4%的条件下反应90 min,脂肪酸的转化率为60.2%[40]。可见,Amberlyst系列催化剂的活性与反应温度及树脂类型直接相关。
Purolite D5081和Purolite D5082是一类新型的具有高交联度的强酸性聚苯乙烯树脂。Abidin等[41]考察了Purolite D5081、Purolite D5082、Amberlyst 36等离子交换树脂催化剂对废弃食用油UCO和甲醇酯化反应的催化活性。Purolite D5081和D5082具有较高的交联度,其催化效果优于具有较低交联度的Amberlyst 36树脂。而Purolite D5081树脂具有较高的比表面积和孔体积,有利于反应物分子与活性位点的接触,显示出较好的催化性能,在反应温度60℃、甲醇/UCO摩尔比6、催化剂用量1.25%、搅拌速率350 r/min的条件下反应8 h,脂肪酸转化率可达88%;Purolite D5081重复使用8次,每轮脂肪酸转化率下降在8%~10%范围。树脂的孔道堵塞以及—SO3H的流失是造成Purolite D5081树脂催化性能下降的主要原因。
强酸性离子交换树脂在生物柴油的制备中体现出较高的催化活性,且易于与产品分离,可循环使用,也不腐蚀设备,但树脂的成本往往比较高,且较高的反应温度往往会导致树脂的失活,在一定程度上也限制了树脂在酸催化领域的应用。因此,继续关注新型树脂的合成对催化反应有很大的意义。
1.4 固体超强酸催化剂
1.5 金属磷酸盐及其有机-无机膦酸盐衍生物
金属磷酸盐也可作为有效的固体酸催化剂应用于生物柴油合成中的酸催化反应中。Das等[46]以F127为模板,在强酸性条件下,采用溶液挥发自组装EISA法合成得到了介孔的含氧磷酸锆材料,并将其作为催化剂用于酯化反应。在醇/酸摩尔比10、催化剂用量5%、反应温度338 K的条件下反应10 h,月桂酸、肉豆蔻酸、棕榈酸和硬脂酸的转化率分别为74.4%、86.41%、75.5%和74.2%。同时,该催化剂重复使用5次,催化活性仍无明显变化,表明该催化剂具有较好的酸催化活性和稳定性。Jiménez-Morales等[47]以十六烷基三甲基溴化铵为表面活性剂合成得到了介孔磷酸钽,并将其作为酸催化剂应用于葵花油与甲醇的酯交换反应中。在醇/油摩尔比12、催化剂用量5%、反应温度200℃的条件下反应2 h,葵花油的转化率达到93%;经3次循环使用,催化剂的活性无明显变化,但第4次循环使用时催化活性开始降低,可能是由于材料表面吸附了中间产物或反应产物所导致。Bassan等[48]考察了磷酸铌对一些长链脂肪酸酯化反应的催化活性。在醇/酸摩尔比10、催化剂用量10%、反应温度120℃的条件下反应7 h,月桂酸、肉豆蔻酸、硬脂酸和油酸的转化率分别为97%、97%、94%和85%;催化剂连续使用3次,其酸催化活性仍无明显损失。Chen等[49]采用水热方法合成了具有三维网络结构的铜钒磷酸盐(CuVOP),并将其作为固体酸催化剂应用于豆油的酯交换反应中。在催化剂用量1.5%、甲醇/油摩尔比6.75、反应温度65℃的条件下反应 5 h,豆油中甘油三酯的转化率可达65.5%。Dutta等[50]以非离子表面活性剂P123为结构导向剂,合成出了具有较大介孔的磷酸锡催化剂 (LPSnP-1),其平均孔径可达10.4 nm。在423 K微波辅助条件下催化果糖、葡萄糖等碳水化合物的脱水反应,果糖、葡萄糖、蔗糖、纤维二糖和纤维素生成5-羟基甲基糠醛的产率分别为77%、50%、51%、39%和32%。而且该催化剂重复使用5次后,产率只下降4%,表明其具有较好的催化活性和稳定性,也显示其在制备生物柴油等酸催化反应中的应用潜力。
有机膦酸盐作为无机磷酸盐的衍生物,除具有无机磷酸盐的一些优点,如较好的稳定性、酸性、多相性等,又引入了丰富的有机官能团,能够有效地调整材料表面的亲、疏水性,易对其进行表面修饰等。此外,与有机硅烷试剂相比,有机膦酸及其衍生物具有更加丰富的种类和有机官能团,合成步骤简单,同时其成本比较低,因此,有机无机杂合膦酸盐作为一种新型材料日益受到研究者们的关注,并且在吸附分离、生物传感和药物控释以及催化等领域得到应用[51-62]。
有机膦酸盐材料自身即存在一定的酸性,这一酸性主要来源于未与金属螯合的P—OH缺陷位和金属的路易斯酸性[63-64],因此,有机无机杂合膦酸盐本身可作为酸催化剂。Dutta等[65]以植酸为有机膦源,以SnCl4·5H2O为无机前驱体,采用无模板溶胶-凝胶方法合成了具有结晶孔墙的有机-无机杂合介孔膦酸锡块体MLSnP-1,并作为固体酸催化剂催化一系列长链脂肪酸和甲醇的酯化反应。在323 K下经过6 h反应,脂肪酸甲酯的产率高于93%;在常温下反应24 h,月桂酸甲酯的产率可达94%。MLSnP-1在生物柴油制备中表现出较高的催化活性,可能是由于其具有一定的质子酸位点P—OH以及一定的路易斯酸性。该催化剂重复使用5次,催化活性仍无明显降低,显示出较好的稳定性。
Pramanik等[66]以苯基-1,3,5-三膦酸为有机膦源,氯化铁为金属源,在酸性介质(pH=4)、无模板剂条件下水热合成了一种结晶的、超微孔的有机膦酸铁纳米材料HFeP-1-3,并将其用于氰乙酸乙酯的酯交换反应。在催化剂用量1%、反应温度60℃的温和条件下反应6 h,转化率高达88.3%。HFeP-1-3对该反应较高的催化活性主要取决于其中的Fe活性位点以及氰乙酸乙酯中CN较强的吸电子效应,使反应物与活性位点易于接触。在催化剂重复使用5次后,氰乙酸甲酯的产率从88.9%下降为85.6%,HFeP-1-3仍能保持较高的催化活性。但该催化剂主要适用于具有较少流动性电子的酯的酯交换反应,在生物柴油制备中受到一定的应用限制。
有机-无机杂合金属膦酸盐作为固体酸催化剂时,未与金属螯合的P—OH缺陷位是其酸性的主要来源之一。但是,在传统的合成方法中,绝大多数磷羟基与金属中心发生配位,从而使该缺陷位浓度较低,所得材料的酸量较少。因此,采用新型的方法来合成具有较高P—OH浓度的有机膦酸盐催化剂对于酸催化反应很重要。在一系列烷基胺的存在下,Ma等[67]合成了一种具有高酸量的介孔膦酸钛材料,其合成路径如图2所示。其中,烷基胺作为P—OH缺陷的保护基团,先通过酸碱反应占据一定的P—OH位点,这就在一定程度上减少了P—OH与金属中心的络合;之后,再经过HCl萃取处理,又将保护起来的P—OH缺陷位释放出来,从而有效地提高了金属膦酸盐中P—OH的浓度。采用这种方法得到的有机无机金属膦酸盐的H+交换量为5.76 mmol/g,明显高于不经烷基胺保护合成得到的膦酸钛的酸量2.54 mmol/g。有机金属膦酸盐和纯无机磷酸盐具有相似的孔结构、酸量和酸强度,因此前者具有更高催化活性的原因可能是杂合膦酸盐材料中存在疏水性有机基团,使材料表面与反应底物存在更强的亲和力[63,67]。这种新型固体酸催化剂也具有较好的稳定性,可应用于制备生物柴油的酸催化反应中。
由于有机膦酸的种类丰富,从而使金属膦酸盐中含有的有机基团不同,有利于进一步功能化。如经磺化处理后引入磺酸基团(见图3)[63],可以有效提高材料的酸强度和酸催化活性。以HEDP为有机膦源合成的有序介孔膦酸钛PMTP-2,再经ClSO3H磺化处理,其H+离子交换容量可从3.93 mmol/g提高到6.62 mmol/g。以磺化后PMTP-2催化油酸和甲醇的酯化反应,在75℃下反应5 h,油酸转化率达87.3%,相较于采用PMTP-2(4.9%)时有显著提高。磺化后的PMTP-2在经过80℃热水处理后仍保持着强酸性质,即磺酸基在较高温度下也能较稳定地保留在材料的孔墙中,在一定程度上说明了该催化剂具有较好的稳定性。另外,含苯环的有机膦酸作为配体合成的介孔膦酸锌HZnP-1,经浓硫酸磺化后得到的磺化膦酸锌催化剂HZnPS-1[68],用其催化一系列长链脂肪酸和甲醇的酯化反应,在反应温度298 K、催化剂用量5%条件下反应24 h,脂肪酸转化率均高达95%,且在重复使用5次后,仍具有较高的催化活性。
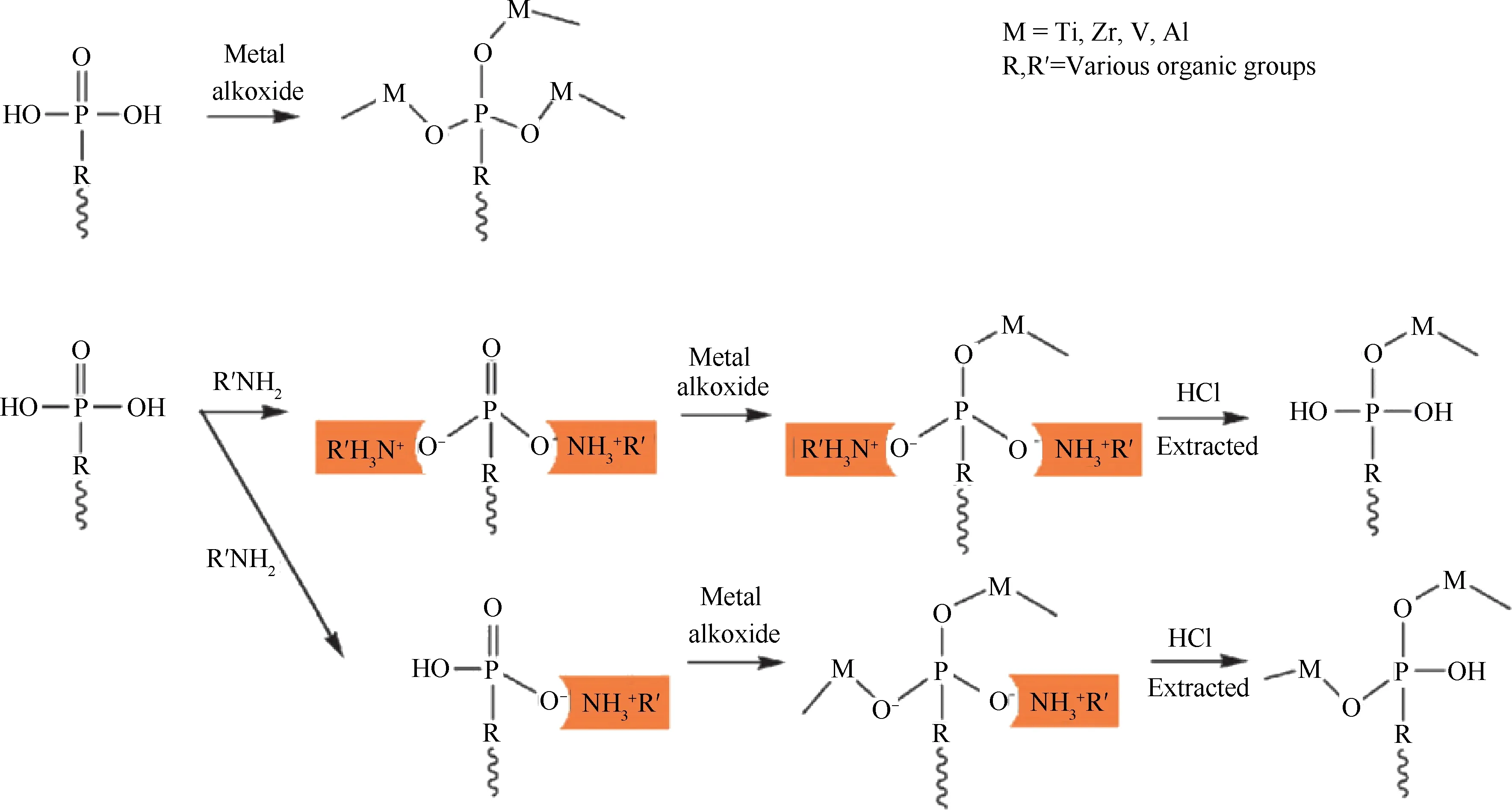
图2 烷基胺辅助合成高酸性有机膦酸钛材料示意图
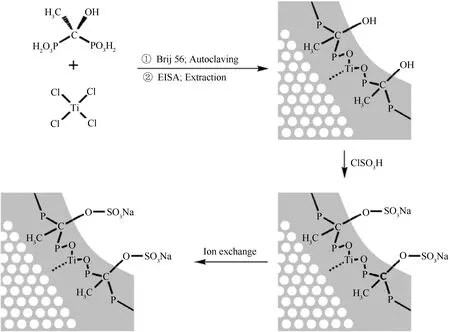
图3 PMTP-2的合成、硫酸酯化和离子交换过程
有机无机杂合金属膦酸盐具有一定的质子酸和路易斯酸酸性[69-70],且可将其中的有机官能团进一步功能化,应用于制备生物柴油的酸催化反应中。但是,合成高度有序的膦酸盐材料仍然存在一定的难度,且由于其本身缺乏P—OH质子酸酸量和酸强度,酯化反应往往需要在较高的醇/酸摩尔比下进行。因此,通过改变合成条件、选择合适的膦酸配体及表面修饰方法来进一步提高新型杂合膦酸盐固体酸催化剂的酸量和酸强度,仍是研究者需要努力的一个方向。
1.6 碳基固体酸催化剂
碳基固体酸催化剂是一种新型固体酸催化剂,不仅可以有效避免酸腐蚀及产生大量废水的不利环境的因素,显著降低生产成本,而且具有热稳定性高、催化活性高、重复使用性好等优点,近年来受到研究者们的广泛关注。
1.6.1 介孔碳基固体酸
介孔碳材料由于其具有较好的化学稳定性、较高的比表面积、可调节的孔道结构、疏水性的表面、表面易被—SO3H和—COOH修饰等优点,被广泛地作为催化剂载体应用于工业和环境保护等领域[71],在生物柴油制备中也得到了应用。
Janaun等[72]以SiO2为硬模板,D-葡萄糖为碳前驱体,发烟硫酸为磺化试剂,制备了介孔碳固体酸催化剂,将其应用于催化油酸和甲醇的酯化反应,并考察了SiO2模板对所制备催化剂的物理化学性质及其酸催化活性的影响。在SiO2模板除去前后分别对材料进行磺化处理,在磺化处理后再除去硬模板所得的CMK-SO3H-w呈现出较好的介孔结构,具有较高的比表面积和孔体积;而先除去硬模板再进行磺化处理所得的CMK-w-SO3H的比表面积和孔体积急剧下降,表明在磺化过程中其介孔结构的坍塌(见图4)。以前者的处理方法,SiO2模板的存在能够作为支撑物使介孔碳材料的介孔结构在磺化处理过程中得到较好的保持,但SiO2的存在也在一定程度上阻碍了—SO3H对材料内表面的修饰,从而使
CMK-SO3H-w具有较低的酸量;以后者的处理方法,所得CMK-w-SO3H比CMK-SO3H-w具有更高的总酸量和—SO3H含量,在油酸和甲醇的酯化反应中表现出更高的催化活性。对于油酸和甲醇的酯化反应,相对于催化剂的比表面积,催化剂的总酸量尤其是—SO3H的密度是更为重要的决定性因素。Kitano等[73]在研究磺化多孔碳催化剂催化乙酸的酯化反应中也得到了相似的结论。然而,合成出同时具有较好的孔道结构、较高的比表面积和较高的酸含量的碳基催化剂仍是一个挑战,对进一步提高碳基固体酸催化剂在制备生物柴油的酸催化反应中的应用具有更大的实际意义。
以表面活性剂(如三嵌段共聚物F127和P123等)作为模板,酚类化合物(如间苯三酚、间苯二酚和苯酚等)和甲醛的聚合物作为碳的前驱体,经有机-有机自组装作用能合成得到不同结构的介孔碳材料[74-76]。Chang等[77]以F127为模板,间苯二酚和甲醛作为碳的前驱体,采用软模板法合成得到介孔碳材料,经过碳化和磺化处理后,制备出一系列磺酸化介孔碳基固体酸催化剂。在较低的碳化温度(400℃)下得到的磺化介孔碳催化剂具有最高的酸量,这可能是由于较低的碳化温度有利于保存介孔碳材料表面的—OH,促进—SO3H基团对介孔碳材料表面的修饰,从而使所得MC-4-SO3H具有较高的酸含量,在油酸和甲醇的酯化反应中表现出最高的催化活性;采用0.1 g MC-4-SO3H催化剂,在甲醇/油酸摩尔比30、反应温度70℃的条件下反应3 h,油酸的转化率可以达到95%以上。该催化剂在重复使用5次后,油酸的转化率仍能达到90%。
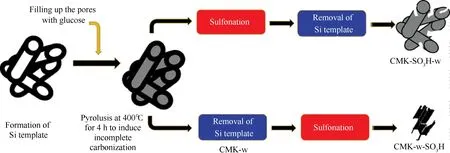
图4 制备CMK-w-SO3H和CMK-SO3H-w的示意图
1.6.2 生物质碳基固体酸
采用模板法合成的活性炭材料在生物柴油制备中表现出了较高的催化活性,但合成过程比较复杂,成本较高,不适合用于生物柴油的工业化生产。而生物质材料成本较低,普遍存在,并且在低温碳化处理后富有表面基团,易于对其进行后修饰,近年来以生物质材料作为原料制备固体酸催化剂应用于制备生物柴油,受到了广泛关注。
以碳水化合物(如D-葡萄糖、蔗糖、纤维素、淀粉等)为原料制备的固体酸催化剂用于制备生物柴油,比一些传统的固体酸催化剂,如沸石、铌酸、大孔树脂等,表现出更高的催化活性[78-80]。以淀粉为原料,在300℃下碳化后得到的活性炭材料,经浓硫酸或浓硫酸和氯磺酸混合液作为磺化试剂处理后,比表面积明显下降,表明磺酸基成功嫁接到该碳材料表面,而总酸量可高达10 mmol H+/g,在催化油酸和乙醇的酯化反应中表现出较高的活性[81]。在反应温度80℃、油酸/醇摩尔比1/10,催化剂用量1.33%条件下反应25 h,油酸转化率达92%。但此催化剂在第2轮循环使用时,油酸的转化率降至67%,与其磺酸基含量的降低一致 (磺酸基质量摩尔浓度由2.3 mmol SO3H/g降至1.6 mmol SO3H/g),表明磺酸基的流失是其催化活性降低的主要原因,反映出其稳定性并不太理想。
Nakajima等[82]以微晶纤维素为原料,以发烟硫酸为磺化试剂,制备出固体酸催化剂CCSA-SO3H;在碳化温度低于723 K时催化剂表面存在大量的磺酸基、羧基以及酚羟基(见图5)。在637 K碳化制备的CCSA-SO3H催化剂在催化油酸与甲醇的酯化反应中,比传统的固体酸催化剂如铌酸、磺化的大孔树脂Amberlyst-15、全氟磺酸树脂Nafion NR50等表现出更高的催化活性;在368 K下反应2 h,油酸甲酯的产率已达到90%,达到相同反应条件下浓硫酸活性的70%~80%;反应4 h,油酸甲酯的产率为99.9%。使用过的CCSA-SO3H经水洗和403 K烘干后再次使用,在重复使用10次,催化活性仍无明显下降。CCSA-SO3H对甘油三酯的酯交换反应也表现出较高的催化活性,在温和的反应条件下(反应温度为353 K,反应6 h),油酸甲酯的产率可达24.1%,明显优于其他一些传统的固体酸催化剂(Amberlyst-15为5.0%,Nafion NR50为1.5%,二氧化硅负载的全氟磺酸Nafion SAC-13为1.1%);催化剂重复使用5次,其催化活性仍无明显降低。上述结果表明,CCSA-SO3H这种无定形碳基固体酸催化剂在酯化反应和酯交换反应中具有较高的催化活性和稳定性。
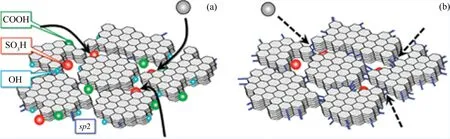
图5 不同碳化温度下合成的SO3H修饰的CCSA材料的结构示意图
对于生物质碳基催化剂,原材料的选择对催化剂的催化活性有着明显的影响。选择废弃生物质日益受到人们的重视,因为不仅能够有效地缓解能源危机,而且也能有力地减少对环境的污染[83-84]。不同国家地区富有的废弃生物质不同,这也就为原材料的选择提供了多样性。如在中国南方地区,制糖工业每年产生大量的甘蔗渣废弃生物质(大约每年800万t)。Lou等[85]以甘蔗渣为原料,经碳化和磺化处理后,制备出富含磺酸基、羧基和羟基的碳基固体酸催化剂,并详细考察了碳化时间、碳化温度、磺化时间以及磺化温度对该催化剂催化酯化反应和酯交换反应活性的影响,也考察了催化剂的物质结构对其催化活性的影响。结果表明,碳化时间对该催化剂的酯化和酯交换催化活性没有明显影响,而磺化时间有一定影响;随着磺化时间的延长,催化剂的活性随之提高。相较于碳化时间和磺化时间,碳化温度和磺化温度对催化剂活性的影响更为重要,如图6所示。由图6(b)可以看出,过低和过高的碳化温度都不利于该催化剂得到较高的催化活性。过低碳化温度所得到的材料中含有较小的多环芳烃,它们在较高温度下容易流失,从而影响催化剂的活性;过高碳化温度所得材料中可能形成较大的多环芳烃层以及碳层的堆积,而磺酸基主要嫁接在碳层的边缘部分[86],在过高温度下所得到的刚性结构也不利于与磺酸基的接触,从而使磺酸基密度比较低,导致其酯化和酯交换反应的活性也比较低;在碳化温度为648 K时,所得材料具有较高的磺酸基密度,其催化活性也最高。由图6(d)可以看出,该催化剂的活性随磺化温度呈现出先升高后降低的趋势,423 K为最佳磺化温度。该催化剂重复使用8次,催化活性仍保持为初始活性的90%以上,表明该催化剂具有较好的稳定性和重复使用性能。相比较而言,大孔树脂Amberlyst-15和铌酸的催化活性明显低于该生物质碳基固体酸催化剂,并且重复使用2~3次,其催化活性明显降低。玉米秸秆[87]、稻壳[88]等作为原料,经合适温度的碳化和磺化处理后制得的生物质碳基固体酸催化剂,对油酸和甲醇的酯化反应也都表现出较高的催化活性。可见,选择合适的生物质材料作为原料,可以制得性能优异的新型固体酸催化剂,对制备生物柴油以及环境保护都有重要的实际意义。
采用模板法合成的介孔碳材料在生物柴油制备中表现出较高的催化活性。但是与生物质原材料相比,合成过程较复杂、成本较高,且生物质材料普遍存在,在低温碳化处理后具有较丰富的表面基团,易对其进行后修饰。因此,利用生物质材料制备固体酸催化剂,在解决能源危机的同时,又重新再利用了废弃生物质。但是,选择合适的生物质作为原材料,同时提高活性组分与基体的相互作用以提高材料的稳定性仍是研究者们需要研究和关注的一个问题。

图6 制备条件对甘蔗渣碳基催化剂的酯化反应(□ 和条纹棒)和酯交换反应(△和黑色棒)催化活性的影响以及不同固体酸催化剂催化油酸和甲醇酯化反应的使用稳定性
2 结论与展望
传统的固体酸催化剂往往具有亲水表面,水或丙三醇等亲水性物质易吸附在催化剂表面,限制了疏水性的反应物与活性位点的接触,同时也会在一定程度上使催化剂失活,降低催化剂的催化活性和重复使用性能,因而,具有疏水性表面的有机-无机杂合材料和碳基固体酸等新型催化剂已倍受关注。在催化剂中引入含有疏水性烷基基团的有机硅烷试剂能够有效地改善催化剂的孔道结构以及表面疏水性,合成得到的硅基有机-无机杂合固体酸催化剂在很大程度上提高了酸催化效率和稳定性。但是,有机硅烷试剂的种类较少,原料不易得,且价格也比较昂贵,在原料选择上具有一定的局限性。相比较而言,有机膦酸及其衍生物具有更加丰富的种类和有机官能团,合成步骤简单,且成本比较低,所合成的相应有机-无机杂合金属膦酸盐具有一定的质子酸和路易斯酸酸性,其中的有机基团能有效改善材料表面的疏水性,能进一步被功能化,因此在酸催化合成生物柴油等反应中有较好的应用前景。但是,合成高度有序的介孔金属膦酸盐材料仍然存在一定的难度,且由于其本身缺乏P—OH质子酸酸量和酸强度,酯化反应往往需要在较高的醇/酸摩尔比下进行,因此,有必要进一步开发提高介孔金属膦酸盐酸量或提高酸强度的新方法和新途径。
生物质原料成本低、来源广,且经过低温碳化处理后易与-SO3H等强酸性基团修饰,可用于生物柴油的制备中。相对于合成的磺酸化介孔碳材料,生物质碳基固体酸催化剂对生物柴油制备反应呈现出相近或更加优异的性能,具有较大的实际应用价值。但是该类型催化剂在使用过程中仍存在活性组分流失等问题,故通过选择合适的原材料和磺化试剂以及改进合成方法等途径来进一步改善材料的稳定性,需要进一步的探索。由此可见,固体酸催化剂在生物柴油制备中仍然存在一定的不足,进一步研究具有较好的孔道结构、高酸量、高酸强度及耐水性等特征的新型固体酸催化剂仍是今后努力的方向。
[1] LUQUE R, LOVETT J C, DATTA B, et al. Biodiesel as feasible petrol fuel replacement: A multidisciplinary overview[J]. Energy & Environmental Science, 2010, 3 (11): 1706-1721.
[2] HUANG Y B, FU Y. Hydrolysis of cellulose to glucose by solid acid catalysts[J]. Green Chemistry, 2013, 15(5): 1095-1111.
[3] SHARMA Y C, SINGH B, KORSTAD J. Advancements in solid acid catalysts for ecofriendly and economically viable synthesis of biodiesel[J]. Biofuels Bioproducts & Biorefining-Biofpr, 2011, 5 (1): 69-92.
[4] LOTERO E, LIU Y, LOPEZ D E, et al. Synthesis of biodiesel via acid catalysis[J]. Industrial & Engineering Chemistry Research, 2005, 44 (14): 5353-5363.
[5] SU F, GUO Y H. Advancements in solid acid catalysts for biodiesel production[J]. Green Chemistry, 2014, 16 (6): 2934-2957.
[6] KINNEY A J, CLEMENTE T E. Modifying soybean oil for enhanced performance in biodiesel blends[J]. Fuel Processing Technology, 2005, 86 (10): 1137-1147.
[7] GRANADOS M L, ZAFRA POVES M D, ALONSO D M, et al. Biodiesel from sunflower oil by using activated calcium oxide[J]. Applied Catalysis B: Environmental, 2007, 73 (3-4): 317-326.
[8] AL-ZUHAIR S. Production of biodiesel: Possibilities and challenges[J]. Biofuels Bioproducts & Biorefining-Biofpr, 2007, 1 (1): 57-66.
[9] SELVABALA V S, SELVARAJ D K, KALIMUTHU J, et al. Two-step biodiesel production from calophyllum inophyllum oil: Optimization of modifiedβ-zeolite catalyzed pre-treatment[J]. Bioresource Technology, 2010, 102 (2):1066-1072.
[10] JACOBSON K, GOPINATH R, MEHER L C, et al. Solid acid catalyzed biodiesel production from waste cooking oil[J]. Applied Catalysis B: Environmental, 2008, 85 (1-2): 86-91.
[11] BUSCA G. Acid catalysts in industrial hydrocarbon chemistry[J]. Chemical Reviews, 2007, 107 (11): 5366-5410.
[12] DI SERIO M, TESSER R, PENGMEI L, et al. Heterogeneous catalysts for biodiesel production[J]. Energy & Fuels, 2008, 22 (1): 207-217.
[13] 张秋云,杨松, 李虎. 制备生物柴油的固体酸催化剂研究进展[J]. 化工进展,2013, 32 (3): 575-583. (ZHANG Qiuyun, YANG Song, LI Hu. Research progress in the preparation of biodiesel with solid acid as the catalyst[J]. Chemical Industry and Engineering Progress, 2013, 32 (3): 575-583.)
[14] SANI Y M, DAUD W M A W, AZIZ A R A. Activity of solid acid catalysts for biodiesel production: A critical review[J]. Applied Catalysis A: General, 2014, 470: 140-161.
[15] JOTHIRAMALINGAM R, WANG M K. Review of recent developments in solid acid, base, and enzyme catalysts (Heterogeneous) for biodiesel production via transesterification[J]. Industrial & Engineering Chemistry Research, 2009, 48 (13): 6162-6172.
[16] BRITO A, BORGES M E, OTERO N. Zeolite Y as a heterogeneous catalyst in biodiesel fuel production from used vegetable oil[J]. Energy & Fuels, 2007, 21 (6): 3280-3283.
[17] KISS A A, DIMIAN A C, ROTHENBERG G. Solid acid catalysts for biodiesel production-towards sustainable energy[J]. Advanced Synthesis & Catalysis, 2006, 348 (1-2): 75-81.
[18] 董松涛, 李宣文,李大东,等. 水热处理USY二次孔形成规律研究[J]. 物理化学学报,2002, 18 (3): 201-206. (DONG Songtao, LI Xuanwen, LI Dadong, et al. Study on the formation of mesopore during hydrothermal dealumination of Y zeolite[J]. Acta Physico-Chimica Sinica, 2002, 18 (3): 201-206.)
[19] 王德举,刘仲能, 李学礼,等. 介孔沸石材料[J]. 化学进展,2008, 20 (5): 637-643. (WANG Deju, LIU Zhongneng, LI Xueli, et al. Mesoporous zeolite materials[J]. Progress in Chemistry, 2008, 20 (5): 637-643.)
[20] BORGES L D, MOURA N N, COSTA A A, et al. Investigation of biodiesel production by HUSY and Ce/HUSY zeolites: Influence of structural and acidity parameters[J]. Applied Catalysis A: General, 2013, 450: 114-119.
[21] ZHANG Q Q, MING W X, MA J H, et al. De novo assembly of a mesoporous beta zeolite with intracrystalline channels and its catalytic performance for biodiesel production[J]. Journal of Materials Chemistry A, 2014, 2 (23): 8712-8718.
[22] 王恩波, 段颖波, 张云峰, 等. 杂多酸催化剂连续法合成乙酸乙酯[J].催化学报, 1993, 14 (2): 147-149.(WANG Enbo, DUAN Yingbo, ZHANG Yunfeng, et al. Continuous process for synthesis of ethyl acetate with heteropoly acids as catalysts[J]. Chinese Journal of Catalysis, 1993, 14 (2): 147-149.)
[23] HU C W, HASHIMOTO M, OKUKARA T, et al. Catalysis by heteropoly compounds XXII reactions of esters and esterification catalyzed by heteropolyacids in a homogenous liquid phase-effect of the central atom of heteropoly anions having tungsten as the addenda atom[J]. Journals of Catalyst, 1993, 143 (2): 437-448.
[24] IZUMI Y, HASEBE R, URABE K. Catalysis by heterogeneous supported heteropoly acids[J]. Journals of Catalyst, 1983, 84 (2): 402-409.
[25] FERNANDES S A, ARDOSO A L, SILVA M J D. A novel kinetic study of H3PW12O40-Catalyzed oleic acid esterification with methanol via1H NMR spectroscopy[J]. Fuel Processing Technology, 2012, 96: 98-103.
[26] BAROI C, DALAI A K. Simultaneous esterification, transesterification and chlorophyll removal from green seed canola oil using solid acid catalysts[J]. Catalysis Today, 2013, 207: 74-85.
[27] KULKARNI M G, GOPINATH R, MEHER L C, et al. Solid acid catalyzed biodiesel production by simultaneous esterification and transesterification [J]. Green Chemistry, 2006, 8 (12): 1056-1062.
[28] SRILATHA K, ISSARIYAKUL T, LINGAIAH N, et al. Efficient esterification and transesterification of used cooking oil using 12-Tungstophosphoric acid (TPA)/ Nb2O5catalyst[J]. Energy & Fuels, 2010, 24 (9): 4748-4755.
[29] ARMATAS G S, BILIS G, LOULOUDI M. Highly ordered mesoporous zirconia-polyoxometalate nanocomposite materials for catalytic oxidation of alkenes[J]. Journal of Materials Chemistry, 2011, 21 (9): 2997-3005.
[30] OLIVEIRA C F, DEZANETI L M, GARCIA F A C, et al. Esterification of oleic acid with ethanol by 12-tungstophosphoric acid supported on zirconia[J]. Applied Catalysis A: General, 2010, 372 (2): 153-161.
[31] BRAHMKHATRI V, PATEL A. Esterification of lauric acid with butanol-1 over H3PW12O40supported on MCM-41[J]. Fuel, 2012, 102: 72-77.
[32] ZHU Y P, REN T Z, YUAN Z Y. Mesoporous non-siliceous inorganic-organic hybrids: A promising platform for designing multifunctional materials[J]. New Journal of Chemistry, 2014, 38 (5): 1905-1922.
[33] SU F, WU Q Y, SONG D Y, et al. Pore morphology-controlled preparation of ZrO2-based hybrid catalysts functionalized by both organosilica moieties and Keggin-type heteropoly acid for the synthesis of levulinate esters[J]. Journal of Materials Chemistry A, 2013, 1 (42): 13209-13221.
[34] DUAN X X, LIU Y, ZHAO Q, et al. Water-tolerant heteropolyacid on magnetic nanoparticles as efficient catalysts for esterification of free fatty acid[J]. RSC Advances, 2013, 3 (33): 13748-13755.
[35] PETERS T A, BENES N E, HOLMEN A, et al. Comparison of commercial solid acid catalysts for the esterification of acetic acid with butanol[J]. Applied Catalysis A: General, 2006, 297 (2): 182-188.
[36] PARK J Y, WANG Z M, KIM D K, et al. Effects of water on the esterification of free fatty acids by acid catalysts[J]. Renewable Energy, 2010, 35 (3): 614-618.
[39] ILGEN O. Investigation of reaction parameters, kinetics and mechanism of oleic acid esterification with methanol by using Amberlyst 46 as a catalyst[J]. Fuel Processing Technology, 2014, 124: 134-139.
[40] GAN S Y, NG H K, CHAN P H, et al. Heterogeneous free fatty acids esterification in waste cooking oil using ion-exchange resins[J]. Fuel Processing Technology, 2012, 102: 67-72.
[41] ABIDIN S Z, HAIGH K F, SAHA B. Esterification of free fatty acids in used cooking oil using ion-exchange resins as catalysts: An eficient pretreatment method for biodiesel feedstock[J]. Industrial & Engineering Chemistry Research, 2012, 51 (45): 14653-14664.
[42] ZHANG Y, WONG W T, YUNG K F. Biodiesel production via esterification of oleic acid catalyzed by chlorosulfonic acid modified zirconia[J]. Applied Energy, 2014, 116: 191-198.
[43] FARCASIU D, LI J Q, CAMERON S. Preparation of sulfated zirconia catalysts with improved control of sulfur content II Effect of sulfur content on physical properties and catalytic activity[J]. Applied Catalysis A: General, 1997, 154 (1-2): 173-184.

[46] DAS S K, BHUNIA M K, SINHA A K, et al. Synthesis, characterization, and biofuel application of mesoporous zirconium oxophosphates[J]. ACS Catalysis, 2011, 1(5): 493-501.
[47] JIMÉNEZ-MORALES I, SANTAMARíA-GONZLEZ J, MAIRELES-TORRES P, et al. Mesoporous tantalum phosphate as acidic catalyst for the methanolysis of sunfiower oil[J]. Applied Catalysis B: Environmental, 2012, 123-124: 316-323.
[48] BASSAN I A L, NASCIMENTO D R, SAN GIL R A S, et al. Esterification of fatty acids with alcohols over niobium phosphate[J]. Fuel Processing Technology, 2013, 106: 619-624.
[49] CHEN L, YIN P, LIU X G, et al. Biodiesel production over copper vanadium phosphate[J]. Energy, 2011, 36 (1): 175-180.
[50] DUTTA A, GUPTA D, PATRA A K, et al. Synthesis of 5-hydroxymethylfurural from carbohydrates using large-pore mesoporous tin phosphate[J]. ChemSusChem, 2014, 7(3): 925-933.
[51] MA T Y, LIN X Z, ZHANG X J, et al. Hierarchical mesostructured titanium phosphonates with unusual uniform lines of macropores[J]. Nanoscale, 2011, 3(4): 1690-1696.
[52] MA T Y, ZHANG X J, YUAN Z Y. Hierarchically meso-/macroporous titanium tetraphosphonate materials: Synthesis, photocatalytic activity and heavy metal ion adsorption[J]. Microporous and Mesoporous Materials, 2009, 123 (1-3): 234-242.
[53] MA T Y, YUAN Z Y. Periodic mesoporous titanium phosphonate spheres for high dispersion of CuO nanoparticles[J]. Dalton Transactions, 2010, 39 (40): 9570-9578.
[54] MA T Y, YUAN Z Y. Metal phosphonate hybrid mesostructures: Environmentally friendly multifunctional materials for clean energy and other applications[J]. ChemSusChem, 2011, 4 (10): 1407-1419.
[55] MA T Y, YUAN Z Y. Organic-additive-assisted synthesis of hierarchically meso-/macroporous titanium phosphonates[J]. European Journal of Inorganic Chemistry, 2010, (19): 2941-2948.
[56] MA T Y, LIN X Z, YUAN Z Y. Periodic mesoporous titanium phosphonate hybrid materials[J]. Journal of Materials Chemistry, 2010, 20 (35): 7406-7415.
[57] ZHU Y P, MA T Y, REN T Z, et al. Mesoporous cerium phosphonate nanostructured hybrid spheres as label-free Hg2+fluorescent probes[J]. ACS Applied Materials & Interfaces, 2014, 6 (18): 16344-16351.
[58] ZHU Y P, MA T Y, LIU Y L, et al. Metal phosphonate hybrid materials: From densely layered to hierarchically nanoporous structures[J]. Inorganic Chemistry Frontiers, 2014, 1 (5): 360-383.
[59] ZHU Y P, LIU Y L, REN T Z, et al. Hollow manganese phosphonate microspheres with hierarchical porosity for efficient adsorption and separation[J]. Nanoscale, 2014, 6 (12): 6627-6636.
[60] ZHU Y P, LIU Y L, REN T Z, et al. Mesoporous nickel phosphate/phosphonate hybrid microspheres with excellent performance for adsorption and catalysis[J]. RSC Advances, 2014, 4 (31): 16018-16021.
[61] ZHU Y P, REN T Z, YUAN Z Y. Hollow cobalt phosphonate spherical hybrid as high-efficiency Fenton catalyst[J]. Nanoscale, 2014, 6(19): 11395-11402.
[62] 刘亚录,朱运培, 李敏,等. 介孔金属膦酸盐杂化材料研究进展[J]. 化学学报,2014, 72(5): 521-536. (LIU Yalu, ZHU Yunpei, LI Min, et al. Advances in mesoporous metal phosphonate hybrid materials[J]. Acta Chimica Sinica, 2014, 72(5): 521-536.)
[63] MA T Y, YUAN Z Y. Functionalized periodic mesoporous titanium phosphonate monoliths with large ion exchange capacity[J]. Chemical Communications, 2010, 46 (13): 2325-2327.
[64] JONES D J, APTEL G, BRANDHORST M, et al. High surface area mesoporous titanium phosphate:Synthesis and surface acidity determination[J]. Journal of Materials Chemistry, 2000, 10 (8): 1957-1963.
[65] DUTTA A, PATRA A K, UYAMA H, et al. Template-free synthesis of a porous organic-inorganic hybrid tin(IV) phosphonate and its high catalytic activity for esterification of free fatty acids[J]. ACS Applied Materials & Interfaces, 2013, 5 (20): 9913-9917.
[66] PRAMANIK M, BHAUMIK A. Organic-inorganic hybrid supermicroporous Iron(III) phosphonate nanoparticles as an efficient catalyst for the synthesis of biofuels[J]. Chemistry-A European Journal, 2013, 19 (26): 8507-8514.
[67] MA T Y, LIU L, DENG Q F, et al. Increasing the H+exchange capacity of porous titanium phosphonate materials by protecting defective P—OH groups[J]. Chemical Communications, 2011,47 (21): 6015-6017.
[68] PRAMANIK M, NANDI M, UYAMA H, et al. Organic-inorganic hybrid porous sulfonated zinc phosphonate material: Efficient catalyst for biodiesel synthesis at room temperature[J]. Green Chemistry, 2012, 14 (8): 2273-2281.
[69] LIN X Z, YUAN Z Y. Synthesis of mesoporous zirconium organophosphonate solid-acid catalysts[J]. European Journal of Inorganic Chemistry, 2012, (16): 2661-2664.
[70] LIN X Z, YUAN Z Y. Synthesis of amorphous porous zirconium phosphonate materials: Tuneable from micropore to mesopore sizes[J]. RSC Advances, 2014, 4 (61): 32443-32450.
[71] MA T Y, LIU L, YUAN Z Y. Direct synthesis of ordered mesoporous carbons[J]. Chemical Society Reviews, 2013, 42 (9): 3977-4003.
[72] JANAUN J, ELLIS N. Role of silica template in the preparation of sulfonated mesoporous carbon catalysts[J]. Applied Catalysis A: General, 2011, 394 (1-2): 25-31.
[73] KITANO M, ARAI K, KODAMA A, et al. Preparation of a sulfonated porous carbon catalyst with high specific surface area[J]. Catalysis Letters, 2009, 131 (1-2): 242-249.
[74] WAN Y, SHI Y, ZHAO D Y. Supramolecular aggregates as templates: Ordered mesoporous polymers and carbons[J]. Chemistry of Materials, 2008, 20 (3): 932-945.
[75] ZHANG F Q, MENG Y, GU D, et al. An aqueous cooperative assembly route to synthesize ordered mesoporous carbons with controlled structures and morphology[J]. Chemistry of Materials, 2006,18 (22): 5279-5288.
[76] LU A H, SPLIETHOFF B, SCHÜTH F. Aqueous synthesis of ordered mesoporous carbon via self-assembly catalyzed by amino acid[J]. Chemistry of Materials, 2008, 20 (16): 5314-5319.
[77] CHANG B B, FU J, TIAN Y L, et al. Soft-template synthesis of sulfonated mesoporous carbon with high catalytic activity for biodiesel production[J]. RSC Advances, 2013, 3 (6): 1987-1994.
[78] TODA M, TAKAGAKI A, OKAMURA M, et al. Green chemistry: Biodiesel made with sugar catalyst[J]. Nature, 2005, 438: 178-178.
[79] LOU W Y, ZONG M H, DUAN Z Q. Efficient production of biodiesel from high free fatty acid-containing waste oils using various carbohydrate-derived solid acid catalysts[J]. Bioresource Technology, 2008, 99 (18): 8752-8758.
[80] ZONG M H, DUAN Z Q, LOU W Y, et al. Preparation of a sugar catalyst and its use for highly efficient production of biodiesel[J]. Green Chemistry, 2007, 9 (5): 434-437.
[81] ALDANA-PÉREZ A, LARTUNDO-ROJAS L, GMEZ R, et al. Sulfonic groups anchored on mesoporous carbon Starbons-300 and its use for the esterification of oleic acid[J]. Fuel, 2012, 100: 128-138.
[82] NAKAJIMA K, HARA M. Amorphous carbon with SO3H groups as a solid Brønsted acid catalyst[J]. ACS Catalysis, 2012, 2(7): 1296-1304.
[83] HUBER G W, IBORRA S, CORMA A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering[J]. Chemical Reviews, 2006, 106 (9): 4044-4098.
[84] RINALDI R, SCHÜTH F. Design of solid catalysts for the conversion of biomass[J]. Energy & Environmental Science, 2009, 2 (6): 610-626.
[85] LOU W Y, GUO Q, CHEN W J, et al. A highly active bagasse-derived solid acid catalyst with properties suitable for production of biodiesel[J]. ChemSusChem, 2012, 5(8): 1533-1541.
[86] OKAMURA M, TAKAGAKI A, TODA M, et al. Acid-catalyzed reactions on flexible polycyclic aromatic carbon in amorphous carbon[J]. Chemistry of Materials, 2006, 18 (13): 3039-3045.
[87] LIU T T, LI Z L, LI W, et al. Preparation and characterization of biomass carbon-based solid acid catalyst for the esterification of oleic acid with methanol[J]. Bioresource Technology, 2013, 133: 618-621.
[88] LI M, CHEN D Y, ZHU X F. Preparation of solid acid catalyst from rice husk char and its catalytic performance in esterification[J]. Chinese Journal of Catalysis, 2013, 34 (9): 1674-1682.
Advances in Solid Acid Catalysts for Biodiesel Production
LIU Yalu,ZHU Yunpei,YUAN Zhongyong
(TianjinCollaborativeInnovationCenterofChemicalScienceandEngineering,KeyLaboratoryofAdvancedEnergyMaterialsChemistryofEducationMinistry,NankaiUniversity,Tianjin300071,China)
biodiesel; solid acid; acid catalysis
2014-12-04
国家自然科学基金项目(21073099)和高等学校博士学科点专项基金项目(20110031110016)资助
刘亚录,女,硕士研究生,从事无机-有机多功能材料合成及应用方面的研究
袁忠勇,男,教授,博士,从事纳米催化材料化学的研究;Tel: 022-23509610; E-mail:zyyuan@nankai.edu.cn
1001-8719(2015)03-0627-16
O643.36
A
10.3969/j.issn.1001-8719.2015.03.004
