ZnO/Al2O3催化对苯二甲酸脱羧反应性能及失活
2015-06-28蒋斌波陆飞鹏廖祖维王靖岱阳永荣黄正梁
蒋斌波,陆飞鹏,庄 岩,廖祖维,王靖岱,阳永荣,黄正梁
(浙江大学 化学工程与生物工程学院 化学工程联合国家重点实验室, 浙江 杭州 310027)
ZnO/Al2O3催化对苯二甲酸脱羧反应性能及失活
蒋斌波,陆飞鹏,庄 岩,廖祖维,王靖岱,阳永荣,黄正梁
(浙江大学 化学工程与生物工程学院 化学工程联合国家重点实验室, 浙江 杭州 310027)
采用共沉淀法制备了ZnO/Al2O3催化剂,并将其应用于对苯二甲酸(TPA)脱羧反应中,采用XRD、TG-DTA、N2吸附-脱附等方法表征制备的催化剂,分别考察了焙烧温度、ZnO负载量、质量空速以及水/TPA摩尔比对TPA脱羧反应的影响,并研究了ZnO/Al2O3催化剂的反应稳定性以及失活机理。结果表明,采用焙烧温度为600℃制备的ZnO负载量(质量分数)为30%的ZnO/Al2O3催化剂,在质量空速0.5 h-1、无水条件下,TPA脱羧的转化率最高,苯收率可达到86%。催化剂表面积炭和尖晶石相晶粒的增长均会导致脱羧催化剂失活。
脱羧;催化;氧化锌;失活;积炭
聚对苯二甲酸乙二酯(PET)是重要的热塑性工程塑料,主要用于生产聚酯薄膜、包装瓶、聚酯纤维等。热解处理技术可以有效地将PET等塑料废弃物转化为可燃低分子化合物,近年来受到国内外普遍关注[1-4]。但PET热解过程中生成大量的对苯二甲酸(TPA),容易造成反应管路和阀门等的腐蚀和堵塞[5-7]。此外,TPA也是PTA(精对苯二甲酸)生产过程中氧化单元排放的固体废弃物(氧化残渣)中有机酸的主要组分[8],其处理和回收再利用较为困难,需要开发新的氧化残渣利用的工艺。
采用催化脱羧的方法将TPA转化为苯等芳烃,不仅可以解决废弃PET塑料热解过程中设备腐蚀的问题,同时为PTA工艺氧化残渣的回收利用开辟了有效途径。但现有的TPA脱羧催化剂存在苯收率低、副产物多、积炭量较高等问题。Masuda等[5]使用FeOOH催化对苯二甲酸脱羧,反应产物主要是二氧化碳、二苯甲酮、苯甲酸等,苯的含量较少。Kumagai等[6-7]考察了TPA在CaO上的催化脱羧行为,苯是主要产物,但CaO变成CaCO3,降低了催化脱羧活性,且积炭量(质量分数)在20%以上。
庄岩等[9]认为,ZnO作为TPA脱羧催化剂具有相比于其他金属氧化物更高的活性和苯的选择性,并且揭示了其脱酸反应的机理。为了进一步提高脱羧反应活性及苯选择性,笔者通过共沉淀法制备了负载型ZnO/Al2O3催化剂,考察了焙烧温度、ZnO负载量、质量空速、水/TPA摩尔比这些因素对其催化脱羧性能的影响,并采用BET、XRD等方法表征了制备的脱羧催化剂,分析催化剂的失活原因。
1 实验部分
1.1 原料
Al(NO)3·9H2O、Zn(NO)2·6H2O、氨水、吡啶和对苯二甲酸,均为分析纯,国药集团化学试剂有限公司产品。
1.2 催化剂制备
采用共沉淀法制备ZnO/Al2O3催化剂。称取一定比例(以催化剂中ZnO质量分数计,分别为0.1、0.2、0.3、0.4以及0.5)的Al(NO3)3·9H2O和Zn(NO3)2·6H2O固体,用去离子水配成一定浓度的水溶液,在60℃下逐滴加入0.1 mol/L的氨水溶液并不断搅拌,直至溶液pH值为9,静置4 h。过滤得到固体沉淀,用去离子水洗涤至中性,在110℃下干燥12 h后,放置在马福炉中于一定温度下焙烧4 h,经研磨、筛分得到40~80目的颗粒备用。焙烧温度为600℃和900℃的ZnO/Al2O3催化剂分别记为Cat-600和Cat-900。
1.3 催化剂表征
采用岛津公司XRD-600型X射线衍射分析仪分析催化剂的物相,管电压40 kV,管电流80 mA,用Cu的Kα射线,连续扫描,扫描角度为5°~90°。通过X-射线宽化法,采用Scherrer公式[10]计算得到催化剂的晶粒大小,如式(1)所示。
d=0.089λ/(B(2θ) cosθ)
(1)
式(1)中,B(2θ)为衍射峰的半峰宽;λ为X射线的波长;θ为衍射峰的位置;d为样品的平均晶粒度。
采用Micromeritics ASAP2020型表面测定仪测定样品比表面积和孔径分布。在测试前,所有样品均在250℃的高真空条件下预处理2 h,然后在液氮条件下(-196℃)测定。按照BJH(Barrett-Joiner-Halenda)模型方法计算得到比表面积和孔径分布。
采用瑞士METTLER TGA/SDTG 851e型热分析仪对反应后催化剂进行失重分析,在30 mL/min空气氛围下,以15℃/min从30℃升至900℃。
1.4 催化剂评价
采用固定床反应器(管径φ25×2.5 mm,催化剂床层高度10 cm)考评脱羧催化剂[9]。催化剂装填量10 g,进料量4.8 g,载气N2流量600 mL/min,反应温度500℃。将TPA溶于吡啶中配制成0.08 g/mL的溶液,以一定的流量通入固定床反应器中,TPA在N2氛围中催化剂的作用下进行脱羧反应,产物冷却后经气、液分离得到气相产物和液相产物。采用在线GC-9860型气相色谱仪(TCD检测器)分析气体产物组成,色谱柱为自制聚合物填充柱,恒温70℃,桥电流120 mA。采用GC-1690型气相色谱仪(FID检测器)分析液体产物组成,DB-5HT毛细管色谱柱(30 m×0.32 mm×0.10 μm),柱温从50℃(5 min)→5℃/min→70℃(1 min)→25℃/min→250℃(3 min)。反应结束后,在N2吹扫下降至室温后,收集催化剂样品,进一步进行表征。
TPA催化脱羧产物各组分质量分数基于进料TPA质量(100%)计算得到,催化剂的积炭残渣质量分数由100%减去脱羧产物组分总质量分数得到。
TPA转化率x(TPA)、苯和CO2收率(y(Benzene),y(CO2))、苯选择性(s(Benzene))、积炭量(w(C))分别由式(2)~(6)计算。
(2)
(3)
(4)
(5)
(6)
式(2)~(6)中,nTPA,0为TPA的总进料量,mol;nTPA,1为TPA实际转化的量,mol;nB为苯的生成量,mol;nCO2为CO2实际生成的量,mol;mC为催化剂的积炭总质量,g;mCat为催化剂总质量,g。
2 结果与讨论
2.1 焙烧温度对ZnO/Al2O3催化剂的影响
选取ZnO负载质量分数30%的ZnO/Al2O3催化剂作为考察对象,考察不同焙烧温度对所制备的ZnO/Al2O3催化剂性质的影响。
图1为不同焙烧温度制备的ZnO/Al2O3催化剂的XRD谱。由图1可见,不同焙烧温度下得到催化剂均检测到完整的ZnAl2O4尖晶石结构特征衍射峰(2θ=36.8°、38.5°、59.3°、68.6°、74.1°)。其中,300℃与600℃焙烧得到的催化剂的ZnAl2O4尖晶石特征衍射峰较弱,说明结晶度较差;焙烧温度高于600℃以后,该特征衍射峰强度随焙烧温度升高而逐渐增强,说明结晶度逐渐增加,且ZnAl2O4晶粒逐渐变大。较高的焙烧温度易使ZnAl2O4微晶聚集、长大,低温下焙烧则不易于ZnAl2O4尖晶石的形成[11-12]。另外,图1中各XRD谱并无明显ZnO的特征衍射峰,说明活性组分ZnO在其中以高分散形式或非晶相形式存在[10]。
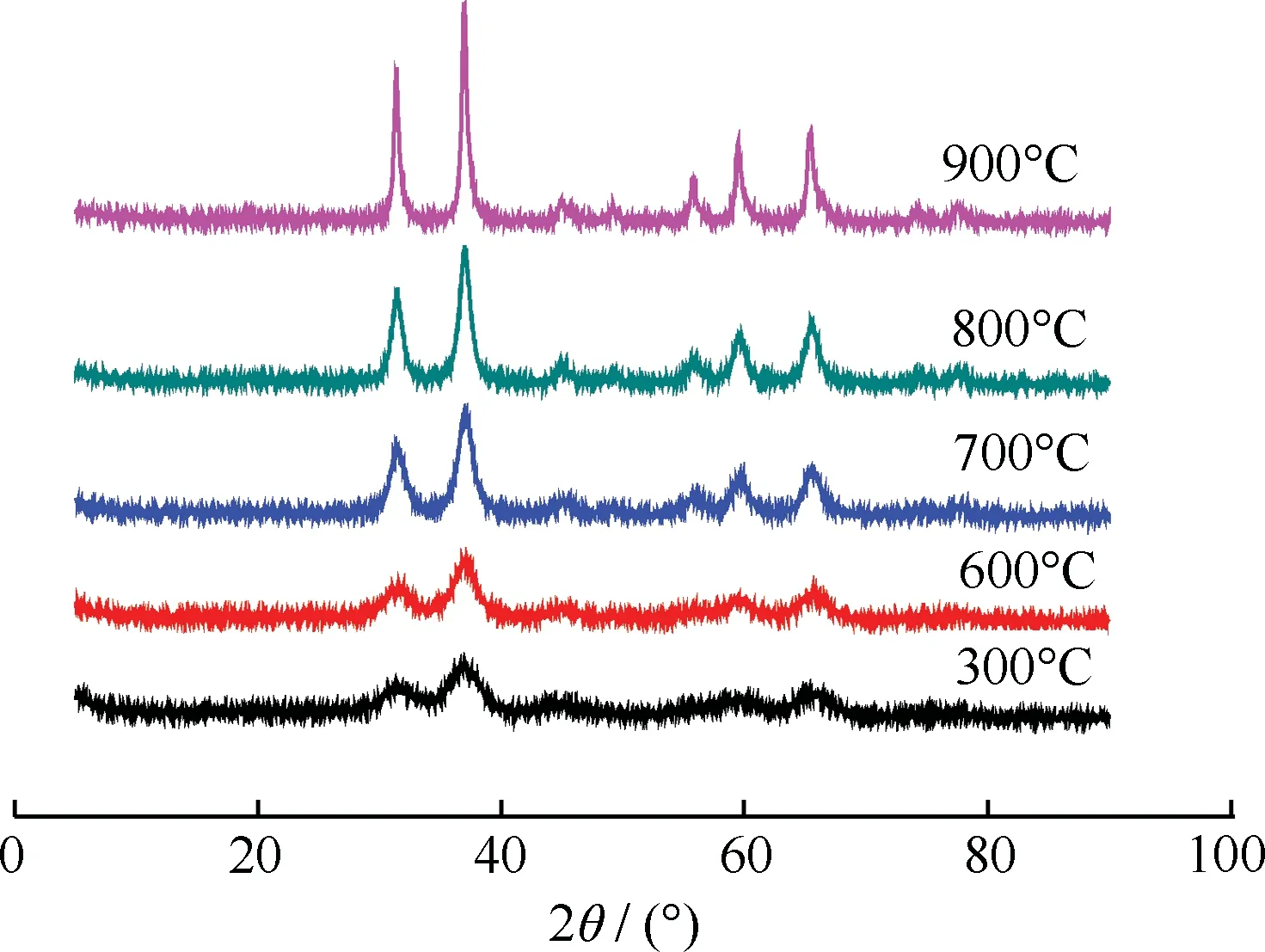
图1 不同焙烧温度制备的ZnO/Al2O3催化剂的XRD谱
表1为ZnO催化剂和焙烧温度为600℃和900℃制备的ZnO/Al2O3催化剂Cat-600、Cat-900催化TPA脱羧反应的产物分布。由表1可知,几种催化剂的脱羧活性和苯的选择性由高到低的顺序均为Cat-600、纯ZnO、Cat-900。随着焙烧温度的升高,活性组分与载体形成更多的ZnAl2O4尖晶石相,当焙烧温度达到900℃时,其活性组分大部分转化为尖晶石相(见图1),因此Cat-900的脱羧活性和苯的选择性均很低。由于脱羧反应在500℃以上进行,因此选择催化剂焙烧温度为600℃。
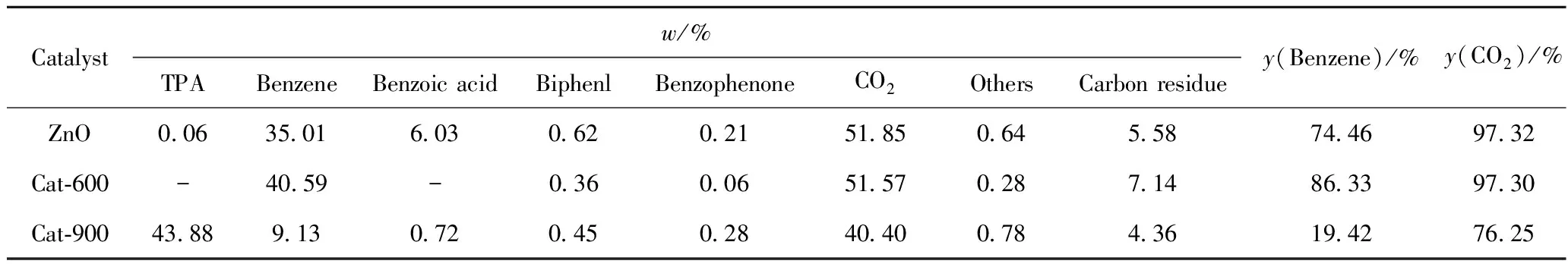
表1 不同焙烧温度制备的ZnO/Al2O3催化TPA脱羧反应的产物分布
p=0.1 MPa;MHSV=0.5 h-1;T=500℃;v(N2)=600 mL/min
2.2 ZnO负载量对ZnO/Al2O3催化TPA脱羧反应的影响
ZnO负载量对ZnO/Al2O3催化TPA脱羧反应的影响示于图2,不同ZnO负载量的ZnO/Al2O3催化剂均在600℃下焙烧得到。从图2可以看出,不同ZnO负载量的ZnO/Al2O3催化TPA脱羧反应的的转化率均接近或等于100%,而产物分布却有较大差异。随着ZnO负载量的增加,产物中苯和二氧化碳的含量不断增加,而催化剂上碳质残渣含量逐渐降低。当ZnO负载质量分数在20%以上时,苯的选择性与纯ZnO催化剂相当,表明负载型ZnO/Al2O3可以促进活性组分在载体表面分散,在保持相同反应活性下可降低催化剂中活性组分含量。当负载质量分数为30%时,TPA转化率为100%,催化剂积炭量达到相对稳定的值。因此,在本研究中以ZnO负载质量分数为30%的ZnO/Al2O3为催化剂,进一步考察其催化TPA脱羧反应的性能。
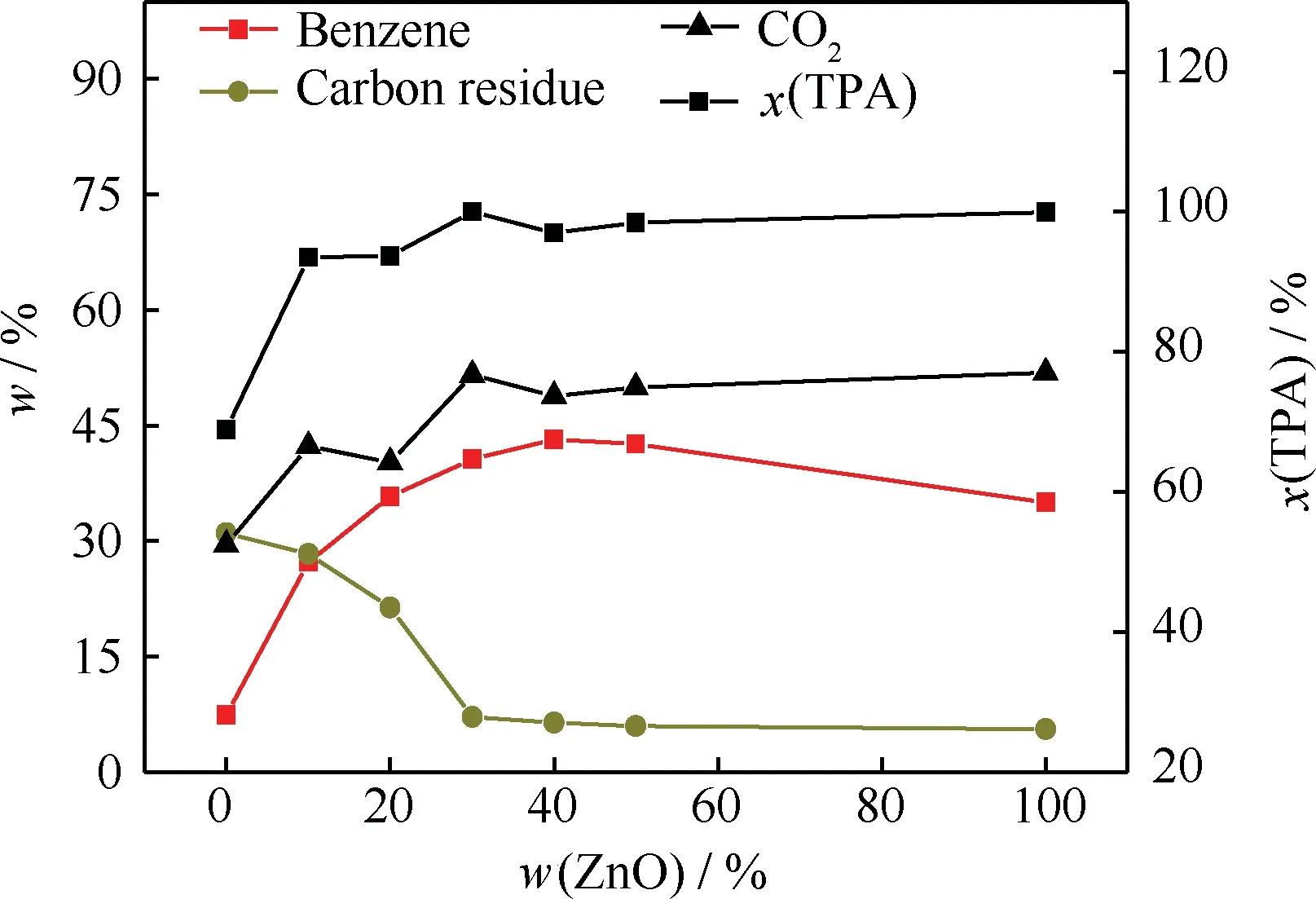
图2 不同ZnO负载量ZnO/Al2O3催化剂催化TPA脱羧反应的结果
2.3 质量空速对ZnO/Al2O3催化TPA脱羧反应的影响
在固定反应温度和进料流量的条件下,通过改变催化剂用量调节反应空速,考察其对ZnO/Al2O3催化TPA脱羧反应的影响,结果示于图3和图4。
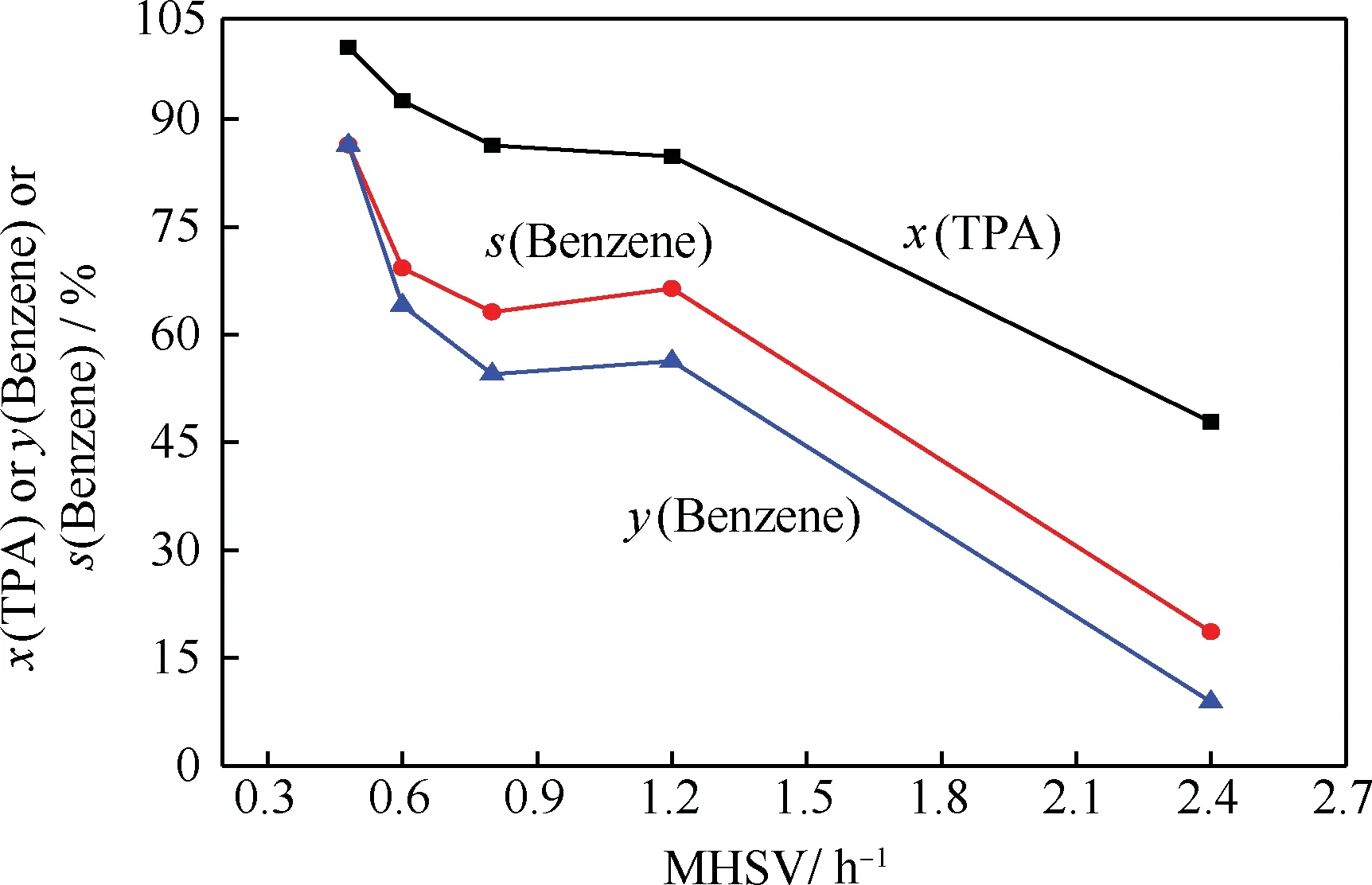
图3 质量空速对ZnO/Al2O3催化TPA脱羧反应的转化率、苯的收率和选择性的影响
从图3和图4可以看出,随着MHSV的减小,TPA在ZnO/Al2O3催化剂层的停留时间增加,催化脱羧反应深度加深,苯的选择性和收率逐渐增加,而产物中苯甲酸含量逐渐降低,说明对苯二甲酸的脱羧历程是串联反应,苯甲酸作为中间产物可进一步脱羧生成苯,因此,在较高的MHSV下,苯甲酸在产物中的含量较高。随着MHSV减小到0.5 h-1以上时,TPA基本完全转化,中间产物苯甲酸也全部参与脱羧反应,此时苯的收率可达到86%左右。TPA的催化脱羧反应可能的反应历程如图5所示。TPA分子首先在催化剂表面吸附反应脱去1个羧基生成苯甲酸,然后苯甲酸作为反应物继续脱羧反应,生成产物苯及二氧化碳。该反应规律与ZnO催化TPA脱羧反应规律[9]一致。

图4 质量空速对ZnO/Al2O3催化TPA脱羧反应产物分布的影响
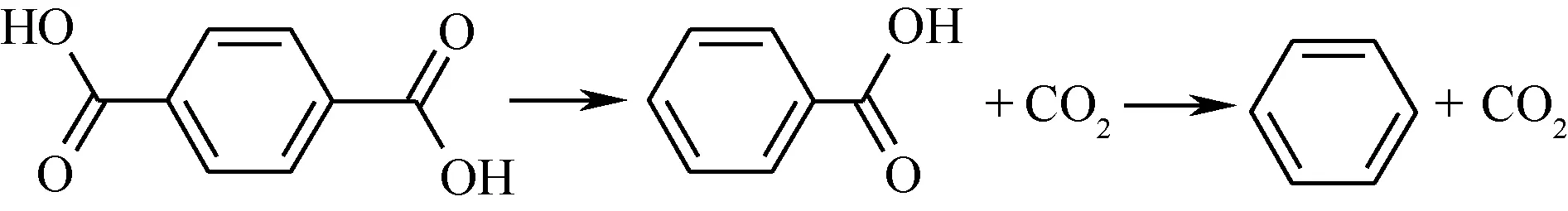
图5 TPA的脱羧串联反应示意图
2.4 水/TPA摩尔比对ZnO/Al2O3催化TPA脱羧反应的影响
在脱羧反应过程中,ZnO/Al2O3催化剂表面易积炭。积炭会覆盖催化剂表面或者堵塞孔道,导致催化剂失活。如果反应条件有利于积炭的气化或抑制反应生成积炭中间物种,就会延长催化剂寿命。
图6和图7分别为水/TPA摩尔比对ZnO/Al2O3催化TPA脱羧反应物转化率、目标产物选择性以及产物分布的影响。可以看出,在相同TPA反应空速下,随着反应物中水/TPA摩尔比的增加,TPA转化率逐渐降低,苯的收率也明显下降。这一方面是因为水与TPA会发生竞争吸附,从而导致转化率有明显的下降;另一方面,根据氧化锌脱羧反应机理[9],如图8所示,对苯二甲酸在脱羧反应过程中首先会与活性碱中心生成羧酸盐配合物,而反应中水的加入会导致羧酸盐中间产物的水解,从而导致苯的选择性显著降低。不过,水的引入可以降低催化剂积炭量(如图7),而且水/TPA摩尔比的增加能够明显抑制副产物(联苯、苯甲酸等)的生成。在含水的反应体系中,系统中存在着生成焦炭前身物的反应和水蒸气使焦炭前身物气化的过程,这两者之间的平衡对于结焦起重要作用。对苯二甲酸盐中间产物热裂解会产生自由基,自由基之间的相互结合产生联苯等多环芳烃。羧酸盐的深度脱氢反应及大分子稠环芳烃很容易导致积炭的生成[13]。水可以作为供氢剂[14],提供部分的自由氢与苯自由基直接结合生成苯,从而降低了多个苯自由基相互结合的概率,使积炭量大大降低。

图6 水/TPA摩尔比(n(H2O)/n(TPA))对TPA转化率、苯的收率和选择性的影响

图7 水/TPA摩尔比(n(H2O)/n(TPA))对脱羧产物分布的影响

图8 TPA在ZnO催化下的脱羧反应机理
2.5 ZnO/Al2O3催化剂失活机理
2.5.1 催化反应稳定性
图9为ZnO/Al2O3催化TPA脱羧反应稳定性考评结果。由图9可见,在脱羧反应的最初阶段,苯和CO2的生成量都有明显的上升趋势,可以认为该阶段为催化剂的活化阶段。随着TPA在ZnO/Al2O3催化下脱羧反应的进行,产物苯和二氧化碳的含量缓慢降低,说明催化剂存在缓慢失活。与此同时,副产物苯甲酸的含量逐渐增加,即催化剂对产物苯的选择性降低,说明苯甲酸脱羧的活性中心受到抑制。
2.5.2 反应前后ZnO/Al2O3催化剂的晶相变化
催化TPA脱羧反应前后的ZnO/Al2O3催化剂的XRD谱示于图10。各个样品的所有特征衍射峰与ZnAl2O4标准XRD谱一致。

图9 ZnO/Al2O3 催化TPA脱羧反应的稳定性
从图10可以看出,反应前后ZnO/Al2O3催化剂的XRD谱没有出现新的衍射峰,ZnAl2O4尖晶石相对应特征衍射峰的强度有所增强,表明晶粒尺寸略有增加。通过计算可知,晶粒大小由4.7 nm增长至5.7 nm。以共沉淀法制备Al2O3负载氧化锌催化剂过程中会有ZnAl2O4尖晶石生成,而且在脱羧反应过程中其尖晶石相有所生长,这可能会导致催化剂脱羧活性部分下降。
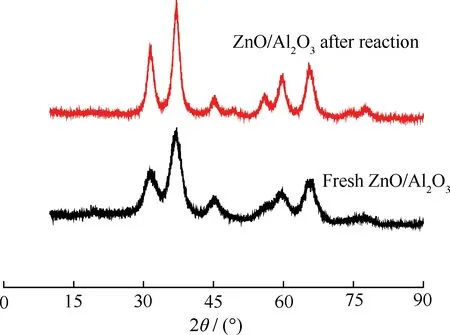
图10 TPA脱羧反应前后ZnO/Al2O3催化剂的XRD谱
2.5.3 反应后催化剂的积炭
为了考察催化剂表面覆盖的积炭情况,对反应420 min后的催化剂样品进行TG-DTA表征,结果示于图11。由图11可以看出,在200℃以下的失重,主要为催化剂上物理吸附水的脱附所致;200~560℃之间有明显的失重,在该温度区间有明显的DTA放热峰出现,因此可以判断该峰为ZnO/Al2O3催化剂上积炭燃烧引起,到560℃左右积炭基本燃烧完毕。由此可以得知,随着反应进行,ZnO/Al2O3催化剂有明显的积炭出现,由TG失重曲线计算可得反应420 min后催化剂积炭量为7.4%。

图11 TPA脱羧反应后ZnO/Al2O3催化剂的TG-DTA曲线
2.5.4 反应前后催化剂的比表面积和孔结构
进一步对TPA脱羧反应420 min后ZnO/Al2O3催化剂比表面积和孔结构进行分析,并以Cat-900作为比较,它们的N2吸附-脱附等温线示于图12。可以看出,Cat-600和Cat-900催化剂的N2吸附-脱附等温线都属于典型的IV型吸附等温线,催化剂对N2吸附量随分压(p/p0)的变化中,在中高压段滞后环非常明显,在低压滞后环闭合,这是典型介孔材料所具有的特征。低压段吸附量平缓增加,此时N2分子以单层到多层吸附于孔内表面。中高压段的突跃位置与催化剂的孔径大小有关,曲线中的突跃段幅度又可以提供孔均匀性的信息,变化幅度大,且斜率高,愈显示孔的均一性。具体参数列于表2。
从表2可见,新鲜Cat-600的比表面积为202.3 m2/g,随着脱羧反应的不断进行,催化剂不断烧结,同时反应过程生成积炭堵塞催化剂内的孔道,二者协同作用导致比表面积下降至147.8 m2/g。而Cat-900催化剂比表面积却由53.2 m2/g增加到98.2 m2/g,孔道内径则由11.9 nm降低到5.4 nm。这表明催化剂上的积炭可能为相对疏松的多孔结构,其孔径大小接近于4 nm,从而可以解释催化剂积炭使得Cat-600的孔径分布变窄,而Cat-900的孔径分布曲线呈现双峰分布的现象。
BET的分析结果表明,反应形成的积炭易堵塞催化剂孔道,导致催化剂平均孔径减小,积炭的形成也导致Cat-600催化剂比表面积下降,也是催化剂失活的一个重要原因。
综上所述,催化剂活性的下降是多种因素共同作用的结果。TPA在发生脱羧反应时会形成一系列的积炭前驱物,随着反应的进行会沉积在催化剂表面和孔道内,覆盖了催化剂的活性点;同时,反应过程中催化剂中ZnAl2O4尖晶石相晶粒生长,也会导致活性中心数量减少,最终表现为催化剂的失活。
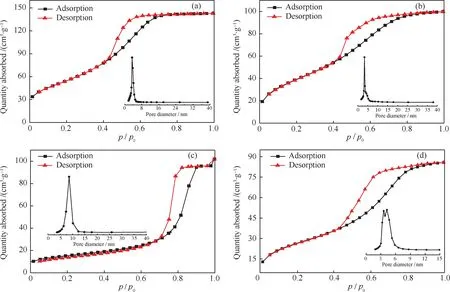
图12 ZnO/Al2O3催化剂的N2吸附-脱附等温线及孔径分布曲线(77 K)
表2 TPA脱羧反应前后ZnO/Al2O3催化剂样品比表面积(SBET)和平均孔径(D)
Table 2SBETandDof ZnO/Al2O3catalyst samples before and after TPA decarboxylation

CatalystSBET/(m2·g-1)D/nmFreshAfterreactionFreshAfterreactionCat⁃600202 3147 84 44 2Cat⁃90053 298 211 95 4
3 结 论
(1) 采用共沉淀法制备的负载型ZnO/Al2O3催化剂,相比于纯ZnO催化剂,其催化TPA脱羧反应的转化率和对产物苯选择性均有所提高。采用600℃焙烧温度制备的ZnO负载质量分数为30%的ZnO/Al2O3催化剂,在反应温度500℃、反应器质量空速0.5 h-1,以及无水的条件下,TPA能够完全转化,苯收率达到86%。
(2) 水作为供氢剂,可以抑制ZnO/Al2O3催化剂积炭。水/TPA摩尔比提高可降低催化剂上积炭的形成,但水不仅会与TPA发生竞争吸附,而且导致催化脱羧反应的羧酸盐中间物的水解,从而降低TPA转化率和苯选择性。
(3) 随着ZnO/Al2O3催化TPA反应的进行,苯的选择性逐渐降低,催化剂存在缓慢失活现象。催化剂积炭和ZnAl2O4尖晶石相晶粒的生长是导致催化剂失活的原因。
[1] MASUDA T, MIWA Y, TAMAGAWA A, et al. Degradation of waste poly(ethylene terephthalate) in a steam atmosphere to recover terephthalic acid and to minimize carbonaceous residue[J]. Polymer Degradation and Stability, 1997, 58(3): 315-320.
[2] YOSHIOKA T, KITAGAWA E, MIZOGUCHI T, et al. High selective conversion of poly(ethylene terephthalate) into oil using Ca(OH)2[J]. Chemistry Letters, 2004, 33(3): 282-283.
[3] TERAKADO O, HIRASAWA M. Effect of metal oxides on the pyrolysis residues of poly(ethylene terephthalate): Formation of carbonaceous submicron, nano-scale filaments and mesoporous compounds[J]. Journal of Analytical and Applied Pyrolysis, 2005, 73(2): 248-256.
[4] TERAKADO O, UEDA M, HIRASAWA M. Thermal degradation of polyester-metal oxide mixtures: Fibrous morphology of carbonaceous compounds and pyrolysis products distribution[J]. Journal of Analytical and Applied Pyrolysis, 2010, 89(2): 183-190.
[5] MASUDA T, MIWA Y, HASHIMOTO K, et al. Recovery of oil from waste poly(ethylene terephthalate) without producing any sublimate materials[J]. Polymer Degradation and Stability, 1998, 61(2): 217-224.
[6] KUMAGAI S, GRAUSE G, KAMEDA T, et al. Decomposition of gaseous terephthalic acid in the presence of CaO[J]. Industrial & Engineering Chemistry Research, 2011, 50(4): 1831-1836.
[7] KUMAGAI S, GRAUSE G, KAMEDA T, et al. Improvement of the benzene yield during pyrolysis of terephthalicacid using a CaO fixed-bed reactor[J]. Industrial & Engineering Chemistry Research, 2011, 50(11): 6594-6600.
[8] 郭霞. 中纯度对苯二甲酸残渣回收方法的研究[D]. 杭州:浙江大学,2009.
[9] 庄岩,蒋斌波, 王靖岱,等. ZnO催化对苯二甲酸脱羧制苯反应历程的研究 [J].石油学报(石油加工), 2015,31(3):698-704. (ZHUANG Yan, JIANG Binbo, WANG Jingdai, et al.Catalytic decarboxylation mechanism of terephthalic acid to benzene using ZnO catalyst[J]. Acta Petrolei Sinica (Petroleum Processing Section),2015,31(3):698-704.)
[10] BORCHERT H, SHEVCHENKO E V, ROBERT A, et al. Determination of nanocrystal sizes: A comparison of TEM, SAXS, and XRD studies of highly monodisperse CoPt3particles[J]. Langmuir, 2005, 21(5): 1931-1936.
[11] 张欣,徐广通, 邹亢,等. S Zorb吸附剂中锌铝尖晶石形成原因的研究[J]. 石油学报(石油加工),2012, 28(2): 242-247. (ZHANG Xin, XU Guangtong, ZOU Kang, et al. Formation mechanism of gahnite in S Zorb sorbents[J]. Acta Petrolei Sinica (Petroleum Processing Section),2012, 28(2): 242-247.)
[12] DEWING E W, ROLSETH S, STØEN L, et al. The solubility of ZnO and ZnAl2O4in cryolitemelts[J]. Metallurgical and Materials Transactions, 1997, 28(6): 1099-1101.
[13] ARTOK L, SCHOBERT H H. Reaction of carboxylic acids under coal liquefaction conditions 1 Under nitrogen atmosphere[J]. Journal of Analytical and Applied Pyrolysis, 2000, 54(1): 215-233.
[14] POLSHETTIWAR V, VARMA R S. Green chemistry by nano-catalysis[J]. Green Chemistry, 2010, 12(5): 743-754.
Reaction Performance and Deactivation of ZnO/Al2O3During the Decarboxylation of Terepthalic Acid
JIANG Binbo, LU Feipeng,ZHUANG Yan, LIAO Zuwei, WANG Jingdai, YANG Yongrong, HUANG Zhengliang
(StateKeyLaboratoryofChemicalEngineering,CollegeofChemicalandBiologicalEngineering,ZhejiangUniversity,Hangzhou310027,China)
ZnO/Al2O3catalyst was prepared by chemical co-precipitation method and then applied in the decarboxylation of terephthalic acid(TPA). The prepared ZnO/Al2O3catalyst was characterized by XRD, TG-DTA and N2adsorption-desorption. The influences of ZnO loading amount, calcination temperature, space velocity and molar ratio of water to TPA on the decarboxylation performance of ZnO/Al2O3were investigated, respectively. The activity stability and deactivation mechanism of the as-prepared ZnO/Al2O3catalyst were also discussed. The results showed that over the ZnO/Al2O3catalyst with ZnO loading amount of 30% and calcined at 600℃, under the MHSV of 0.5 h-1and without water, the highest conversion of the TPA decarboxylation was obtained with the benzene yield of 86%. The carbonaceous deposition and growth of spinel phase would both cause the deactivation of ZnO/Al2O3decarboxylation catalyst.
decarboxylation; catalyze; ZnO; deactivation; carbonaceous deposition
2014-11-24
国家自然科学基金项目(21176208)和国家高技术研究发展计划项目(2012AA030304)资助
蒋斌波,男,副教授,博士,从事工程催化、多相流反应工程研究;Tel:0571-87952254;E-mail:jiangbb@zju.edu.cn
黄正梁,男,助理研究员,博士,从事多相流检测与信息处理研究;Tel:0571-87951227;E-mail:huangzhengl@zju.edu.cn
1001-8719(2015)03-0603-08
TQ032
A
10.3969/j.issn.1001-8719.2015.03.001
