马来酸氯苯那敏的荷移分光光度测定方法
2015-06-05孙海冰余兰
孙海冰 余兰▲
遵义医学院药学院,贵州遵义563000
马来酸氯苯那敏的荷移分光光度测定方法
孙海冰 余兰▲
遵义医学院药学院,贵州遵义563000
目的建立快速便捷测定马来酸氯苯那敏片剂含量的新方法。方法采用分光光度法研究2,3-二氯-5,6-二氰-1,4-苯醌(DDQ)与马来酸氯苯那敏之间的电荷转移反应。结果荷移反应生成的络合物在588 nm有最大吸收波长,表观摩尔吸光系数为3.9×103L/(mol·cm),药物质量浓度在10~100mg/L范围内服从比尔定律,回收率分别为99.0%、98.8%、99.5%,相对标准偏差分别为0.37%、0.54%、0.39%。结论应用本方法测定片剂中马来酸氯苯那敏,精密度好、准确度高,含量测定结果与药典基本一致。
马来酸氯苯那敏;2,3-二氯-5,6-二氰-1,4-苯醌;荷移反应;分光光度法
马来酸氯苯那敏(chlorphenamine maleate)又称扑尔敏,是第一代抗组胺类药物,具有镇静和抗胆碱作用。临床上主要用于治疗各种过敏性疾病,是抗感冒类药物中常见组分。目前已报道的定量测定方法主要有紫外可见分光光度法[1-2]、高效液相色谱[3-7]、气相色谱法[8]、近红外光谱法[9]、毛细管电泳法[10-12]、化学发光[13]、共振光散射光谱[14]和荷移分光光度法[15]等,而以2,3-二氯-5,6-二氰-1,4-苯醌(DDQ)作为电子受体与马来酸氯苯那敏反应还未见报道。本文研究并优化了反应条件,测定了络合物组成,初探了反应机制,建立了马来酸氯苯那敏片剂含量测定新方法。
1 仪器与试药
T9CS分光光度计,北京普析通用;AL204电子天平,梅特勒托利多(上海)有限公司;HWCL-3磁力搅拌反应浴,郑州长城科工贸有限公司。
马来酸氯苯那敏(中国食品药品检定研究院):精密称定马来酸氯苯那敏对照品0.012 50 g,置于25 ml容量瓶中,用乙腈溶解稀释至刻度,得0.5 g/L乙腈溶液;DDQ(上海晶纯生化科技股份有限公司):精密称取DDQ 0.5000 g,置于250ml容量瓶中,用乙腈溶解稀释至刻度,得2mg/ml乙腈溶液;乙腈(色谱纯,Fishercheical公司);乙醇、甲醇、三氯甲烷、丙酮,所有试剂均为分析纯(成都市科龙化工试剂厂);马来酸氯苯那敏片剂(130801,山东仁和堂药业股份有限公司;140513,河南九势制药股份有限公司;140115大同市云岗制药有限公司,规格均为4 mg/片),实验用水为二次去离子水(18.25MΩ·cm)。
2 方法与结果
2.1 样品溶液制备
取同一批号(140115)马来酸氯苯那敏10片,准确称量,研磨混匀后准确称取样品总量的3/5,加适量乙腈,微热搅拌,使其充分溶解,冷却至室温后过滤,滤液用乙腈定容于50 ml容量瓶中(马来酸氯苯那敏浓度约为0.5 g/L)。
2.2 实验步骤
取适量马来酸氯苯那敏溶液(≤1.0mg)于10ml比色管中,加入2.00 ml DDQ溶液,用乙腈稀释至刻度,于60℃下反应20 min。冷却至室温,以试剂空白为参比,在588 nm处测量其吸光度值。
2.3 吸收光谱
按实验方法配制溶液后在T9CS分光光度计上于300~800 nm范围内扫描,并绘制吸收光谱图(图1)。实验表明,在此波长范围内马来酸氯苯那敏无吸收,DDQ最大吸收峰在378 nm,而荷移络合物产生3个吸收峰,分别在458、548、588 nm,最大吸光度产生于588 nm处,所以选此波长作为实验测定波长。

图1 DDQ、马来酸氯苯那敏及络合物的吸收光谱图
2.4 温度与时间对荷移反应的影响
分别在不同温度(25、30、40、50、60℃)下,考察时间对反应体系的影响,测定其吸光度值,结果见表1。
由表1可知,马来酸氯苯那敏与DDQ络合物在25、30、40℃的吸光度变化缓慢,而50、60℃下,反应均快速、完全,且在60℃时仅经20 min,吸光度即达到最大,故实验选择在60℃水浴中放置20min。
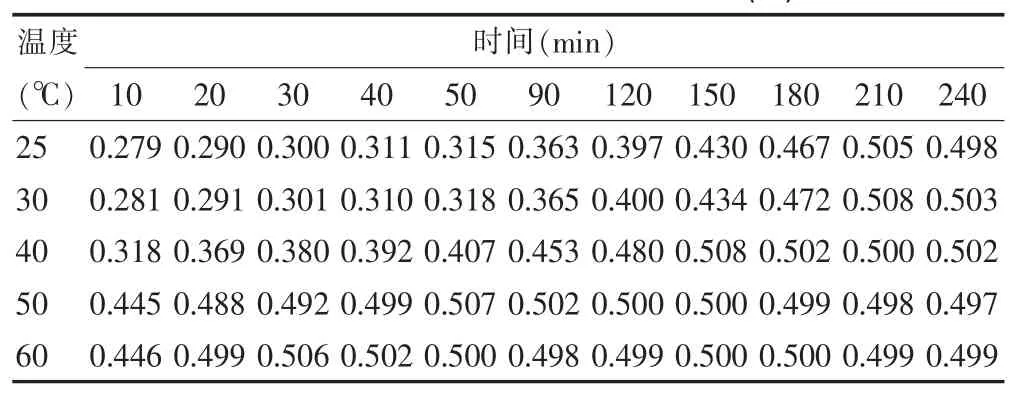
表1 温度与时间对荷移反应的影响(A)
2.5 DDQ用量对荷移反应的影响
取1 ml马来酸氯苯那敏标准溶液于10 ml比色管中,依次改变加入DDQ用量,分别测定其吸光度值,见图2。结果表明,DDQ的最佳用量为2ml。

图2 DDQ用量对荷移反应的影响
2.6 溶剂种类对荷移反应的影响
分别考察了水、甲醇、乙醇、丙酮、乙腈、三氯甲烷作为稀释溶剂对络合物吸光度的影响。按照实验方法,测定的其吸收光度值,结果表明,用乙腈作溶剂时,灵敏度最高。
2.7 马来酸氯苯那敏标准工作曲线的绘制
准确移去不同量的马来酸氯苯那敏标准溶液,在确立的最佳实验条件下进行反应,测定络合物的吸光度,并以马来酸氯苯那敏的质量浓度为横坐标,吸光度值为纵坐标绘制工作曲线(图3)。马来酸氯苯那敏在10~100 mg/L范围内服从比尔定律,线性回归方程为A=0.0101C+0.0189,r=0.9997,检出限为5 mg/L,表观摩尔吸收系数为3.9×103L/(mol·cm)。

图3 马来酸氯苯那敏的标准工作曲线
2.8 方法精密度试验
精密吸取标准溶液1 ml,按照“2.2”项下的方法操作,连续测定6次。测得吸光度分为0.531、0.531、0.532、0.532、0.532、0.533。通过计算,马来酸氯苯那敏平均吸光度是0.532,相对标准偏差(RSD)为0.7%,RSD值较小,说明仪器精密度良好。
2.9 络合物组成及稳定常数测定
用等摩尔连续变化法测定马来酸氯苯那敏与DDQ络合物的组成,结果如图4所示。由图可知,等摩尔连续变化法切线交点大约在0.5,即CM/C=0.5,由此可以推出,络合物组成比为1∶1。根据曲线上最大吸光度与其理论吸光度不同,计算求得稳定常数是2.11×104。

图4 等摩尔连续变化法测定络合物组成
2.10 干扰物对荷移反应的影响
考察了常用片剂中常见辅料对含量测定影响。当马来酸氯苯那敏浓度是1.28×10-4mol/L,相对误差小于±5%时,500倍的葡萄糖、淀粉、蔗糖,100倍的果糖、硬脂酸镁,均对反应无影响。
2.11 试样测定
取0.4 ml样品溶液(浓度约0.5 g/L)于10 ml比色管中,加入2mlDDQ溶液在60℃水浴中反应20min,冷却至室温后在最大波长处进行测定,平行测定5次。测定3个不同厂家马来酸氯苯那敏片剂含量,另按照中国药典方法[1]测定马来酸氯苯那敏片剂含量,并作回收率试验结果分别列于表2、3。

表2 样品中马来酸氯苯那敏的测定(n=5)

表3 回收率试验测定结果(n=6)
2.12 反应机制
马来酸氯苯那敏分子的嘧啶环上氮原子含有一对孤对电子,可以作为电子供体,而DDQ是一个强平面型电子受体,两者可以形成n-π配合物。根据测物质组成测定结果可知,马来酸氯苯那敏与DDQ化学反应计量比是1∶1,由此可推出荷移反应方程式如图5所示。

图5 荷移络合物的形成过程
3 讨论
本实验是基于马来酸氯苯那敏与DDQ之间的电荷转移反应而建立的一种测定马来酸氯苯那敏含量的新方法。实验过程中注意稀释溶剂及温度的选择,不同溶剂体系灵敏度会有很大差别,较高的温度可以使反应较快速达到平衡。
结果表明,马来酸氯苯那敏和DDQ以乙腈作为稀释溶剂,在60℃水浴中,反应20min,形成1∶1红褐色络合物。应用该方法对马来酸氯苯那敏剂含量测定结果与药典基本一致,重现性好,表明可以作为该药物片剂的含量测定质量控制方法。
[1]卫生部药典委员会.中华人民共和国药典·二部[M].北京:化学工业出版社,2010:159-160.
[2]薛晖,费寿耆.系数倍率法测定马来酸氯苯那敏乳膏的含量[J].海峡药学,2004,16(4):16-17.
[3]陈乃江,王统康,张宏.HPLC法测定鼻炎康片中马来酸氯苯那敏的含量[J].浙江中医药大学学报,2010,34(4):600-601.
[4]李桃,林焕泽,吴秀荣.RP-HPLC法测定马来酸氯苯那敏片中马来酸氯苯那敏的含量[J].临床合理用药,2010,3(5):50-51.
[5]马春梅,赵变,席荣英,等.高效液相色谱法测定马来酸氯苯那敏[J].理化检验化学分册,2012,48(10):1244-1246.
[6]杨艳模,石笑弋,易必新,等.高效液相色谱法测定马来酸氯苯那敏糖浆中马来酸氯苯那敏的含量[J].中国药物经济学,2013,8(5):29-31.
[7]沈素梅,唐秀玲,周梁.高效液相色谱法测定维C银翘胶囊中马来酸氯苯那敏的含量[J].临床合理用药,2013,6(7B):30-31.
[8]李丹,常威.气相色谱法测定马来酸氯苯那敏乳膏的含量[J].中国药师,2013,16(1):80-82.
[9]赵吉平,马则秀.近红外光谱法快速检测咳特灵胶囊中马来酸氯苯那敏的含量[J].中国药事,2013,27(2):196-198.
[10]宋粉云,毋福海,钟兆健,等.咳特灵胶囊中马来酸氯苯那敏的高效毛细管电泳-电导法测定[J].中国医药工业杂志,2003,34(6):292-293.
[11]孙元喜,周谷珍,胡霞,等.利用碳纳米管修饰石墨电极测定马来酸氯苯那敏[J].药物分析,2009,29(9):1558-1561.
[12]汪敬武,彭志兵,杨佳.毛细管电泳-电致化学发光法测定维C银翘片中的马来酸氯苯那敏[J].分析试验室,2007,26(4):25-29.
[13]陈小利,马红燕,李超维.反向流动注射化学发光法测定马来酸氯苯那敏[J].分析试验室,2010,29(9):23-25.
[14]时惠敏,冯素玲.共振光散射光谱法测定马来酸氯苯那敏[J].化学研究与应用,2011,23(6):792-795.
[15]冯宇,赵凤林,童沈阳.马来酸氯本那敏与7,7,8,8-四氰基对二次甲基苯醌的荷移反应[J].2003,31(11):1327-1329.
Charge transfer spectrophotometric determ ination method of chlorpheniram inemaleate
SUN Hai-bing YU Lan▲
School of Pharmacy,ZunyiMedical College in Guizhou Province,Zunyi 563000,China
Objective To create a new method to determine the content of chlorpheniramine maleate tablet fast and conveniently.M ethods Charge transfer reaction between 2,3-dichloro-5,6-dicyano-1,4-benzoquinone(DDQ)and chlorpheniramine maleate was studied by spectrophotometry.Results Clathrate generated in charge transfer reaction had maximum absorption wavelength in 588 nm.The apparentmolar absorption coefficientwas 3.9×103L/(mol·cm)and drug concentration ranged from 10-100 mg/L complied with Bill law.The recovery rate was 99.0%,98.8%,99.5% respectively,and relative standard deviation was 0.37%,0.54%,0.39%respectively.Conclusion Determination of chlorpheniramine maleate tablet by this method is good in precision and high in accuracy,and outcome of content determination is basically consistentwith that stipulated in pharmacopeia.
Chlorpheniraminemaleate;2,3-Dichloro-5,6-dicyano-1,4-benzoquinone;Charge transfer reaction;Spectrophotometry
R927.2
A
1674-4721(2015)04(c)-0007-04
2015-01-27本文编辑:许俊琴)
遵义医学院博士科研启动基金(ZMKD2013-006)
孙海冰(1988-),女,江苏徐州人,2012级在读硕士研究生,研究方向:天然产物活性成分分离
▲通讯作者:余兰(1971-),女,贵州遵义人,教授,研究方向:药物分析及天然产物活性成分分离
