固相萃取结合液相色谱-串联质谱技术同时测定饲料中氯苯那敏和溴苯那敏含量
2023-01-06索德成肖志明冯玉超
管 鑫 索德成 肖志明 王 石 冯玉超 梁 英 樊 霞*
(1.中国农业科学院农业质量标准与检测技术研究所,北京100081;2.黑龙江八一农垦大学,大庆163000)
氯苯那敏(chlorphenamine),俗称扑尔敏,化学名为3-(4-氯苯基)-N,N-二甲基-3-吡啶-2-基丙-1-胺;溴苯那敏(brompheniramine),化学名为3-(4-溴苯基)-N,N-二甲基-3-吡啶-2-基丙-1-胺,是氯苯那敏的溴代同系物。溴苯那敏与氯苯那敏具有相似结构,二者同为第一代的H1组胺受体拮抗剂[1],功能性质也类似。近50年来,该类药物被用于治疗多种过敏反应,如花粉热、慢性鼻窦炎/鼻炎以及上呼吸道咳嗽综合征[2-3]等。其中,氯苯那敏因与常规局麻药具有相似的化学结构,同样对心血管和中枢神经系统具有相似的毒性,故也可用作局麻药[4]。在畜牧养殖业中,氯苯那敏具有缓解高致病性猪蓝耳病临床症状[5],稳定低致病性禽流感患鸡血液中白细胞、淋巴细胞、血小板数量和红细胞相关指标[6],以及治疗狂犬病免疫过程中免疫犬过敏症状[7]等功效,此外一些研究也发现二者还具有提高鸡等家禽采食量、日增重以及促进动物生长等作用[8-9]。而同时,有大量研究显示,氯苯那敏和溴苯那敏具有一定的潜在毒副作用,服用该类药物可引起嗜睡、呼吸频率降低等症状[10],严重者还可导致急性中毒,甚至死亡[11-12]。动物采食添加了含氯苯那敏和溴苯那敏的饲料后,可使其蓄积在组织中,而人类食用具有氯苯那敏和溴苯那敏残留的动物组织则会产生上述不良反应。因此,迄今为止,氯苯那敏和溴苯那敏尚未被列入饲料中允许使用的药物范围。但在利益驱使下,一些养殖者将其添加在饲料中饲喂动物,以提高动物采食量、增加体重及促进生长[13],给饲料和畜禽产品质量安全带来了一定程度的潜在隐患。
目前,检测氯苯那敏或溴苯那敏的方法主要是液-质联用(LC-MS)法[13-17]、高效液相色谱(HPLC)法[18-24]等。Bachute等[18]建立了反相高效液相色谱法同时测定片剂中4种活性药物含量的方法,其中氯苯那敏加标回收率为98.61%~101.30%,相对标准偏差(RSD)为0.17%;刘凯双等[20]研究复方金刚烷胺氨基比林片中氯苯那敏的测定方法,其回收率为99.42%~101.70%,RSD为0.84%,该方法可用于检测低含量氯苯那敏;郭倩倩等[24]使用高效液相色谱法测定复方锌布颗粒剂中氯苯那敏的含量,其中氯苯那敏的平均回收率为100.1%,RSD为0.49%,该方法定量限为11.80 μg/kg。
现有检测方法涉及的对象以原料药、血液、动物组织为主[25-31],而对于饲料中氯苯那敏和溴苯那敏的定量分析方法鲜有报道,缺少相关的检测方法。由于饲料的复杂基质干扰是影响方法灵敏度和稳定性的关键因素,所以对其进行净化处理是精确测定被测物质含量必不可少的一步。固相萃取(SPE)技术利用固体吸附剂将液体样品中的被测物质吸附,让被测物质与干扰物质分离,然后再用洗脱液洗脱,达到分离和富集被测物质的目的[32]。基于此,本试验系统研究了饲料中氯苯那敏和溴苯那敏提取和净化技术,建立了利用混合型阳离子固相萃取柱(MCX)结合液相色谱-串联质谱(LC-MS/MS)技术测定饲料中氯苯那敏和溴苯那敏的高效、灵敏定量分析方法,为监测饲料中非法添加氯苯那敏和溴苯那敏的行为提供理论参考。
1 材料与方法
1.1 仪器与试剂
AB SCIEX QTRAP®6500+型超高效液相色谱-串联质谱仪(美国SCIEX公司):配备电喷雾电离源(ESI源)。氯苯那敏和溴苯那敏标准品(纯度>98%)购自德国Dr. Ehrenstorfer公司;甲酸(色谱纯,德国Sigma公司);乙腈和甲醇(色谱纯,美国Fisher ChenAlert Guide公司);其他试剂均为分析纯试剂,购自国药集团化学试剂有限公司。试验用水为经Milli-Q纯化制备的一级水(>18.2 MΩ)。0.22 μm尼龙滤膜(上海安谱实验科技股份有限公司);Oasis HLB固相萃取柱、Oasis MCX固相萃取柱、Oasis MAX固相萃取柱(美国Waters公司);C18固相萃取柱(天津博纳艾杰尔科技有限公司)。不同基质饲料样品由国家饲料质量检验检测中心(北京)提供,并按试验设计制备,具有充分的代表性。
1.2 试验方法
1.2.1 标准溶液的配制
1)混合标准储备液:称取氯苯那敏和溴苯那敏标准品各5 mg(精确至0.01 mg),置于10 mL容量瓶中,用乙腈溶解并定容至刻度,制备氯苯那敏和溴苯那敏含量为500 μg/mL的混合标准储备溶液。
2)混合标准中间液:准确移取2.0 mL混合标准储备液于100 mL棕色容量瓶中,用乙腈定容,配制成10 μg/mL的标准中间溶液。
3)标准工作溶液:用0.1%甲酸∶乙腈(90∶10,v/v)溶液逐级稀释,配制成浓度为1.0~100.0 ng/mL的系列标准工作溶液,该标准系列溶液需临用现配。
1.2.2 样品前处理
精确称取饲料样品5 g,加入20 mL 1%甲酸乙腈溶液,涡旋振荡提取30 min,于10 000 r/min条件下离心5 min,取上清液备用。MCX固相萃取柱依次用3 mL甲醇和3 mL水活化,然后准确移取上清液5 mL过柱,之后依次加入3 mL水和3 mL甲醇淋洗,抽干,最后用5%氨水甲醇溶液3 mL洗脱,收集洗脱液,于40 ℃下氮气吹干。准确加入1 mL 0.1%甲酸∶乙腈(90∶10,v/v)溶液溶解残余物,混匀后过0.22 μm滤膜,上液相色谱-串联质谱仪测定。
1.3 色谱和质谱条件
色谱柱:Waters BEH C18柱(100 mm×3.0 mm, 1.7 μm);柱温:40 ℃;进样量:5 μL;流动相流速:0.4 mL/min,流动相组成及洗脱梯度见表1。
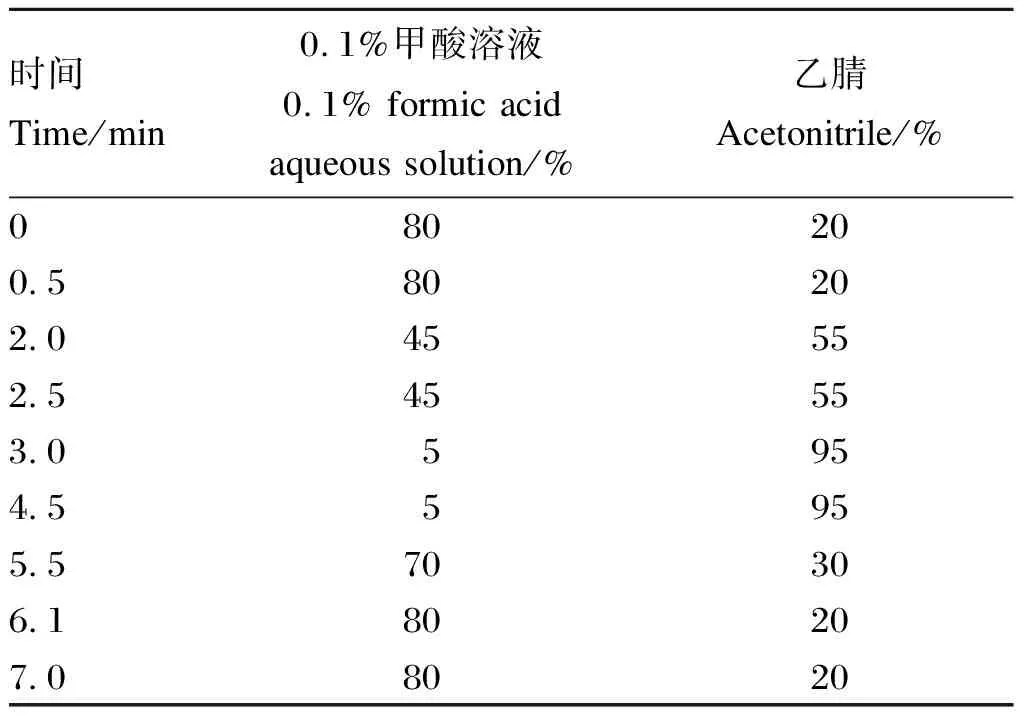
表1 流动相梯度洗脱条件
质谱采用电喷雾电离源(ESI+),多反应监测(MRM)模式;喷雾电压为3.2 kV;离子源温度为350 ℃;气帘气为空气、碰撞气为氮气,使用前调节各气体流量以使质谱灵敏度达到检测要求,分段采集,溴苯那敏和氯苯那敏质谱条件见表2。

表2 溴苯那敏和氯苯那敏质谱条件
2 结果与分析
2.1 质谱条件优化
配制100 ng/mL氯苯那敏和溴苯那敏标准溶液,以微流量泵直接进质谱,进行质谱条件的优化。由于正离子比负离子响应强度高,所以采用ESI+模式的测定。通过一级全扫描质谱得到氯苯那敏和溴苯那敏的母离子[M+H]+为质荷比(m/z)275.2、m/z 319.1;通过子离子扫描得到二级质谱,分别选择m/z较大、强度较稳定的2组m/z 167.2、m/z 230.2(氯苯那敏)和m/z 274、m/z 167.1(溴苯那敏)共4个碎片作为子离子;其中子碎片275.2/167.2(氯苯那敏)、319.1/274.0(溴苯那敏)的响应强度最强且背景干扰小,所以选为定量离子对,另2个子碎片275.2/230.2(氯苯那敏)、319.1/167.1(溴苯那敏)组成定性离子对。在MRM模式下,进一步优化了上述2对离子对的去簇电压(DP)和碰撞电压(CE),结果见表2。
2.2 色谱条件优化
本研究通过对出峰时间、分离效果、峰形和灵敏度等因素分别考察了水(A)-乙腈(B)、0.1%甲酸溶液(A)-乙腈(B)、5 mmol/L甲酸铵(A)-乙腈(B)以及5 mmol/L甲酸铵-0.1%甲酸溶液(A)-乙腈(B)作为流动相时的梯度洗脱效果。结果显示:在相同的梯度洗脱条件下,0.1%甲酸溶液(A)-乙腈(B)作为流动相时,采用0.4 mL/min的流速,洗脱效果更好,具体流动相组成及洗脱梯度见表1。在上述条件下,氯苯那敏和溴苯那敏在该色谱柱上的保留时间分别为2.10、2.25 min,氯苯那敏和溴苯那敏MRM色谱图如图1所示。
2.3 提取试剂选择
试验以配合饲料为对象,研究了1%甲酸乙腈溶液、甲醇∶乙腈∶水(10∶80∶10,v/v/v)溶液、1%盐酸溶液、乙腈、甲醇等5种提取溶剂对氯苯那敏和溴苯那敏的提取效果,称取10份5 g饲料样品,分别加入5种提取剂20 mL,按1.2.2的步骤操作,结果见图2。
从图2可以看出,选取1%甲酸乙腈溶液和乙腈作为提取溶剂对饲料中的氯苯那敏和溴苯那敏具有很好的提取效果,两者的提取效率在90%~95%。而甲醇∶乙腈∶水(10∶80∶10,v/v/v)溶液、1%盐酸溶液、甲醇的提取效率较低,通过对检测结果进行计算,回收率均在60%以下,说明饲料中的氯苯那敏和溴苯那敏提取不完全。
2.4 固相萃取柱的优化
2.4.1 固相萃取柱的选择
试验比较了4种市售的HLB、MCX、MAX、C18固相萃取柱对饲料中氯苯那敏和溴苯那敏的净化效果。先对4种固相萃取柱进行活化处理:加入3 mL甲醇,待甲醇流出后,再加入3 mL水。然后将提取离心后的上清液加入固相萃取柱中,每个小柱10 mL,待样液全部流出后,依次加入3 mL水和甲醇淋洗固相萃取柱,最后MCX用3 mL 5%氨水甲醇溶液洗脱,MAX用3 mL 1%甲酸甲醇溶液洗脱,C18、HLB固相萃取柱用3 mL甲醇洗脱,各收集洗脱液氮吹至近干,用0.1%甲酸∶乙腈(90∶10,v/v)溶液溶解定容至1 mL,上机检测,结果见图3。

图1 氯苯那敏和溴苯那MRM色谱图

误差线为3次测量结果的标准偏差。下图同。
从图3可以看出,经过MCX固相萃取柱净化后,饲料中的氯苯那敏和溴苯那敏具有很好的回收率,在100%左右。而HLB、MAX、C18固相萃取柱的回收率较低,回收率都在50%以下,证明其对氯苯那敏和溴苯那敏并未完全吸附。
2.4.2 洗脱试剂的优化
根据前人研究,氨水甲醇是MCX固相萃取柱的最佳洗脱试剂[33]。所以本试验研究了不同比例的氨水甲醇对氯苯那敏和溴苯那敏的洗脱效果,按照上述操作,洗脱时用不同比例的氨水甲醇进行洗脱,最终上机检测,结果见图4。
从图4可以看出,5%氨水甲醇洗脱效果最好,饲料中的氯苯那敏和溴苯那敏具有很好的回收率,在100%左右。甲醇几乎没有洗脱效果,其他比例的氨水甲醇,虽然也有不错的回收率,但效果不如5%氨水甲醇显著。

图3 不同固相萃取柱对净化效果

图4 不同洗脱试剂的洗脱效果
2.4.3 洗脱溶液用量的优化
作为洗脱溶剂,用量的选择是一个关键参数,因此,试验评估了1.0~5.0 mL 5%氨水甲醇溶液对饲料中氯苯那敏和溴苯那敏的洗脱效果。结果见图5。
从图5可以看出,使用1 mL洗脱液就能较好地将氯苯那敏和溴苯那敏淋洗下来,回收率达到82%;2 mL几乎可以将全部氯苯那敏和溴苯那敏淋洗下来,回收率达到96%;3 mL可以将MCX固相萃取柱上的氯苯那敏和溴苯那敏完全洗脱下来。因此,本研究最终选择3 mL为MCX固相萃取柱的洗脱液用量。
2.5 基质效应
由于不同饲料样品的组成十分复杂,LC-MS/MS检测氯苯那敏和溴苯那敏时基质干扰非常明显,所以基质效应的校正必不可少。取不同饲料样品进行前处理(1.2.2),以此基质提取液作稀释液配制1、5、10、20、50、100 ng/mL的基质标准溶液,与直接用0.1%甲酸∶乙腈(90∶10,v/v)溶液配制的标准溶液对比,大部分氯苯那敏和溴苯那敏表现为基质抑制。为消除基质效应,本试验绘制氯苯那敏和溴苯那敏基质效应图进行校正,结果见图6。

图5 不同洗脱体积的洗脱效果
2.6 标准曲线线性方程、方法检出限和定量限
在上述优化条件下,取空白基质溶液将氯苯那敏和溴苯那敏配制成包含定量限在内的由低到高6个浓度的基质标准溶液,其质量浓度分别为1、5、10、20、50、100 ng/mL,经LC-MS/MS测定分析,以定量离子色谱峰面积与基质添加标准溶液浓度作标准曲线。依据定量离子色谱峰的信噪比(S/N)=3为检出限(LOD),S/N=10为定量限(LOQ),得出LOD和LOQ,结果见表3。基质添加标准溶液在1.0~100.0 ng/mL的线性关系良好;方法检出限为0.3 μg/kg,定量限为1 μg/kg,能够满足饲料中氯苯那敏和溴苯那敏含量的分析需要。
2.7 回收率试验和精密度考察
精密称取配合饲料、预混合饲料与浓缩饲料样品各5 g,然后添加不同含量的氯苯那敏和溴苯那敏标准溶液,配制成1、10、100 μg/kg标准的样品,按上述方法进行检测,每个样品6个重复,连续重复3 d,计算平均回收率、批内及批间变异系数。由表4可知,在1、10、100 μg/kg添加浓度下,平均回收率为85.8%~108.4%,RSD小于15%。
2.8 实际样品测定
通过建立的上述方法对市场上采集的30份不同类型的饲料样品进行检测,在30份饲料样品中有2份样品检出氯苯那敏,分别为预混合饲料(2.37×104μg/kg)和配合饲料(12.58 μg/kg),其余饲料样品均未检出氯苯那敏和溴苯那敏。

图6 氯苯那敏和溴苯那敏基质效应图

表3 基质添加标准曲线的线性方程和相关系数

表4 不同饲料中添加氯苯那敏、溴苯那敏的回收率及精密度
3 讨 论
3.1 流动相的选择
以氯苯那敏和溴苯那敏的分离为主要指标,同时兼顾分析检测时间和色谱图基线平稳情况,选用了乙腈、甲醇[28-31]作为流动相,结果发现,甲醇作为有机相出峰时间较晚且峰形拖尾严重,因此选择了乙腈作为流动相;也对不同水相(水、5 mmol/L甲酸铵溶液、5 mmol/L甲酸铵-0.1%甲酸溶液、0.1%甲酸溶液)进行考察,通过不断摸索优化,最终确定本试验的水相为0.1%甲酸溶液。虽然氯苯那敏和溴苯那敏色谱图部分没有完全分离,但由于二者的定量离子对完全不同,因此并不影响对饲料中氯苯那敏和溴苯那敏精确定量分析。
3.2 提取
文献介绍,使用甲醇、乙腈对人血和中成药样品中氯苯那敏和溴苯那敏进行提取具有较高的提取效率[15,27],但对于饲料样品,根据图2结果可知,甲醇对其提取效率较低,回收率在30%以下,出现这种结果的原因可能是饲料基质复杂,甲醇将饲料中的其他干扰物质也过多溶出,使提取液中干扰物质增多,从而影响了氯苯那敏和溴苯那敏的定量检测;同理甲醇∶乙腈∶水(10∶80∶10,v/v/v)溶液可能也使干扰物质增多,最终影响了检测结果;1%盐酸溶液由于其酸性过强,在饲料提取过程中,可对饲料中一些物质的结构造成破环,从而造成干扰物质剧增,严重影响回收率;1%甲酸乙腈溶液和乙腈在对氯苯那敏和溴苯那敏的提取过程中,表现出良好的提取效果,提取效率达90%以上,为最佳提取液。综合分析发现,氯苯那敏和溴苯那敏在弱酸性条件下保留效果较好,且上机检测过程中还可以增强电离信号,故最终选择1%甲酸乙腈溶液作为提取溶剂。
前人研究表明,通过加入缓冲盐来提高分析物的回收率[34-35]。本研究加入不同种类缓冲盐(醋酸钠、无水硫酸钠、硼砂-碳酸钠、磷酸二氢钾)与1%甲酸乙腈溶液共同提取氯苯那敏和溴苯那敏,观察其对回收率的变化,结果发现,和未加入缓冲盐的试验比较,回收率几乎没有明显影响,为了降低对仪器的不利影响,因此没有加入缓冲盐。
3.3 净化
饲料样品成分多种多样,富含大量矿物质、维生素、蛋白质等,选择合适的净化方式是精确定量分析氯苯那敏和溴苯那敏至关重要的一步[36]。SPE是目前较为热门的净化方法,与传统的基质分散固相萃取方法相比,SPE具有净化效果好、回收率高、溶剂消耗量小、操作简单、省时省力等优点[37]。本试验选择了市售的4种固相萃取柱,对氯苯那敏和溴苯那敏进行净化处理,结果见图3,只有MCX对其展现出良好的净化效果。造成HLB、C18、MAX回收率低的原因可能是其对酸性物质有较好的选择性,而氯苯那敏和溴苯那敏呈现弱碱性;MCX是将磺酸基键合在高度交联的PS/DVB表面得到的混合型强阳离子交换吸附剂,有反相和强阳离子交换双重保留性能,对碱性物质氯苯那敏和溴苯那敏有良好的选择性和灵敏度,所以MCX比HLB、C18和MAX表现出了更加优越的分离富集性能,显著降低了基质干扰,因此,本试验选择了对目标氯苯那敏和溴苯那敏具有高效吸附作用的MCX。
为了得到更好的回收率,本试验对洗脱试剂同样进行严谨的优化处理,结果见图4。甲醇在MCX净化过程中对氯苯那敏和溴苯那敏几乎没有洗脱效果,而加入氨水之后,回收率显著提升,究其原因,MCX作为具有反相和强阳离子交换作用的净化柱,氨水提供了强效的阳离子供应,为氯苯那敏和溴苯那敏的洗脱提供了充足的动力。但是过高或者过低浓度的氨水会使其回收率降低,分析原因,过低浓度的氨水不能提供足够的阳离子,导致洗脱不完全;而过高浓度的氨水则是由于其高浓度会影响氯苯那敏和溴苯那敏的稳定性,导致回收率降低。
3.4 方法性能
目前关于饲料中氯苯那敏和溴苯那敏同步检测的研究尚未见报道,由于饲料成分复杂,形式多样,因此,常规的提取净化方法会造成基体干扰,需要进一步优化提取、净化的方法;此外,在检测技术方面,现有的检测技术虽稳定、可靠,但在分析复杂样品时,其灵敏度和选择性还有诸多不足,如郭倩倩等[24]使用HPLC测定氯苯那敏的定量限仅为11.8 μg/kg;谷亦平等[38]使用HPLC测定氯苯那敏的定量限更是达到58.09 μg/kg;甚至如果仅仅依靠LC可能会出现灵敏度低、容易出现假阳性现象。LC-MS/MS选择性和特异性好,检出限低,近年来是研究抗组胺药物的热门方向,因此本试验采用LC-MS/MS进行检测,其方法检出限为0.3 μg/kg,定量限为1.0 μg/kg,更为灵敏;且加标饲料样品中不同添加浓度(1、10、100 μg/kg)氯苯那敏和溴苯那敏的平均回收率为85.8%~108.4%,RSD<15%,这说明本试验所建LC-MS/MS方法与相关报道方法的回收率没有显著差别,在一定水平上满足饲料中非法添加物质检测的回收率要求,表明了新建立的方法具有较高的准确性和精密度。
4 结 论
本试验通过对提取溶剂、净化等前处理条件及LC-MS/MS分析条件的优化,建立了液相色谱-串联质谱同步检测饲料中氯苯那敏和溴苯那敏含量的方法。氯苯那敏和溴苯那敏含量在1.0~100.0 ng/mL线性关系良好,方法检出限为0.3 μg/kg,定量限为1.0 μg/kg,回收率为85.8%~108.4%,RSD小于15%。该方法前处理操作简单,试剂材料消耗量小,灵敏度高,结果稳定准确,在实际应用中更具有优势。
