线粒体相关肾病2例病例报告及文献复习
2015-05-04李国民方晓燕刘海梅翟亦晖吴冰冰刘学光
李国民 孙 利 沈 茜 徐 虹 方晓燕 曹 琦 刘海梅 翟亦晖 吴冰冰刘学光 杨 青
·论著·
线粒体相关肾病2例病例报告及文献复习
李国民1, 5孙 利1,5沈 茜1徐 虹1方晓燕1曹 琦1刘海梅1翟亦晖1吴冰冰2刘学光3杨 青4
目的 总结2例线粒体相关肾病患儿临床特征及基因突变的特点,提高对该病的认识。方法 收集2例线粒体相关肾病患儿的病史特点、肾脏病理、相关实验室检查和家族史等资料。采用外显子捕获的方法对4 000种人类单基因病的相关致病基因进行高通量测序,包括线粒体DNA A3243G等37个基因和ADCK4等13个参与辅酶Q10生物合成的基因,利用生物信息学对测序结果进行分析,用Sanger法对高通量测序结果进行验证,并在家系中进行突变分析。并进行相关文献复习。结果 2例患儿男女各1例。女性患儿11.7岁起病,主要临床表现为蛋白尿和肾功能异常,无肾外症状,肾脏病理为局灶节段性肾小球硬化(FSGS),检测到NPHS1基因已报道的p.E447K和p.G601A杂合突变,ADCK4基因纯合p.D209H错义突变,为新发现的突变。家系突变分析发现,NPHS1基因p.E447K和p.G601A杂合突变均来自父亲,其哥哥也有相同的基因型,其母亲不携带该2个突变;患儿父母和哥哥分别携带p.D209H杂合突变。男性患儿出生后起病,多个系统受累,表现为精神、运动发育落后,心脏和大血管多发畸形,肾病综合征。检测到COQ6基因的纯合p.R360W错义突变,为新发现的突变。家系突变分析显示,患儿父母分别携带杂合p.R360W错义突变。ADCK4基因p.D209H错义突变和COQ6基因p.R360W错义突变经在线软件PolyPhen和SIFT预测为有害性突变,经多物种蛋白序列比对,2个突变位点均具有保守性。结论 2例患儿肾脏表型分别由辅酶Q10合成基因ADCK4和COQ6突变引起的线粒体相关肾病。新发现p.D209H和p.R360W突变分别丰富了ADCK4和COQ6基因突变谱。
线粒体病; 蛋白尿; 肾病综合征;COQ6基因;ADCK4基因
1 病例报告
例1,女,14岁。2014年5月7日因“发现蛋白尿1月余”就诊于复旦大学附属儿科医院(我院),以蛋白尿原因待查收入我院。
患儿1个月前因“白头发增多”就诊当地医院,查尿常规蛋白3+,未见全身水肿,无尿频、尿急和尿痛,无肉眼血尿及少尿,SCr 98.1 μmol·L-1, BUN 6.1 mmol·L-1,内生肌酐清除率74 mL·min-1·s-2,肾脏病理学检查提示“早期硬化肾小球肾炎”,予黄葵胶囊和贝那普利口服,尿蛋白仍3+。
患儿系G2P2,足月顺产,出生体重3 400 g,既往体健,否认食物、药物过敏史。患儿父母体健,非近亲婚配,母亲妊娠史2-0-0-2,患儿有一个哥哥,体健。
入院查体:血压110/70 mmHg,身高152.0 cm(P50),体重44.0 kg(P50),神志清楚,精神可。全身未见水肿。心、肺、腹部和神经系统查体未见异常。
实验室检查:尿沉渣:蛋白2+、RBC 0.41·HP-1、WBC 0.29·HP-1;尿蛋白/CR 2.79,尿钙/CR 0.04,24 h尿蛋白2.87 g。尿微量蛋白A1M 6.12 mg·L-1,A1MU/CR 22.0 mg·g-1,ALBU 741.0 mg·L-1,ALBU/CR 2 667 mg·g-1,IGGU 22.7 mg·L-1,IGGU/CR 81.7 mg·g-1,NAG 5.2 U·L-1,NAG/CR 2.12 U·mol-1,尿转铁蛋白31.9 mg·L-1。血常规、肝功能、血电解质、血脂、免疫球蛋白和补体指标均正常。自身抗体阴性。SCr 74.0 μmol·L-1,BUN 6.6 mmol·L-1,尿酸44 μmol·L-1,甲状旁腺素182 pg·mL-1。血气分析:pH 7.270、BE -4.2 mmol·L-1。
调阅患儿外院的肾活检病理切片,我院行重新阅片:光镜12个肾小球,7个球性硬化,1个局灶节段性肾小球硬化,4个肾小球正常,明显肾小管萎缩及间质纤维化,肾血管无异常(图1A~C);电镜下足细胞足突部分融合(图1D);免疫荧光显微镜显示(图1E~H):系膜区有IgM、IgA、IgG、C1q和C3沉积(+~++)。 DTPA示双肾灌注欠佳,功能受损,排泄延迟,双肾肾小球滤过率(GFR,未标化)48.4 mL·min-1(左肾26.5、右肾21.9),肾脏MR示左肾实质多发性异常信号,髓质海绵肾待排除,右肾偏小伴右肾可疑异常信号。肾脏磁共振水成像(MRU)和膀胱逆行造影(MCU)未见异常。
诊断:局灶节段性肾小球硬化(FSGS)、慢性肾脏病2期。
因家长拒绝糖皮质激素治疗,继续予贝那普利和和黄葵胶囊口服治疗。
例2,男,10月龄。2015年4月因“心脏杂音,肌张力低下”就诊并收入我院。
患儿出生后因“呕吐14 h”入住当地医院,诊断“新生儿不全性肠梗阻、败血症、先天性心脏病、新生儿肺炎、新生儿窒息、新生儿脑病、肝功能异常、鞘膜积液和足月小样儿”,经治疗好转出院(具体治疗不详)。1个月前因“右侧腹股沟复发性包块8个月”再就诊当地医院,查体发现生长发育落后、左眼睑下垂和四肢肌张力低下;头颅MRI示双侧大脑萎缩,行右侧腹股沟嵌顿性斜疝复位和疝囊高位结扎术。患儿因精神、运动发育落后来我院就诊,心脏超声示:肺动脉狭窄、动脉导管未闭、房间隔缺损、降主动脉流速增快、永存左上腔静脉。
患儿系G1P1,足月顺产,出生体重1 700 g,血性羊水,轻度窒息。生长发育迟缓,目前不能抬头、不能坐;父母亲体健,非近亲婚配,母亲妊娠史1-0-0-1,家族成员否认遗传性疾病史。
入院查体:血压88/50 mmHg,身高65.0 cm(P25),体重5.0 kg (P25),神志清楚,精神、反应欠佳,左侧上眼睑下垂,全身未见明显水肿。HR 130·min-1,律齐,心前区可闻及收缩期Ⅲ/6杂音,以胸骨左缘第2、3肋间最为明显;腹软,无压痛及反跳痛;四肢肌张力低下,不能竖头,不能坐立,克氏、布氏征均阴性。
辅助检查: 尿沉渣:蛋白3+、RBC 0~1·HP-1,WBC 2~3·HP-1;24 h尿蛋白1.04 g ;血常规:Hb 85 g·L-1,RBC 2.64×1012·L-1;血生化:ALT 24 U·L-1,AST 43 U·L-1,总蛋白44.8 g·L-1,白蛋白22.5 g·L-1,总胆固醇5.43 mmol·L-1,三酰甘油2.38 mmol·L-1,SCr 14.0 μmol·L-1,BUN 4.2 mmol·L-1,尿酸218.0 μmol·L-1。血电解质正常。TORCH抗体均阴性,梅毒、HIV和乙肝血清学检查均阴性。免疫球蛋白和补体均正常。血气分析:pH 7.496,BE -0.5 mmol·L-1。泌尿系统B超检查未有异常发现。
诊断:先天性心脏病,肾病综合征,先天性脑发育不良、左侧上眼睑下垂。
行心导管造影和经皮动脉导管未闭封堵术,术后予阿司匹林、呋塞米和螺内酯口服。

图1 例1肾组织病理学检查
Fig 1 Histological changes of kidney in case 1
Notes A-C displayed several sclerosis glomeruli by HE, PAS and PASM staining under light microscope (×200); D showed some fusion of foot processes by electron microscope (×7 000); E-H showed IgG, IgM, C1q and C3 deposition (×200) under immunofluorescence microscope, respectively
2 测序分析
2.1 高通量测序 使用Hiseq 2500高通量模式行PE100测序,按Hiseq 2500标准流程进行。通过Q30的标准对原始数据进行数据过滤得到cleandata和高质量的测序结果。使用bwa比对软件,使用samtool和pindel分析软件得到最初的raw突变;然后经过重复区域、ssr、突变质量值、突变深度、突变率等严格过滤,得到高度可靠的突变,在dbSNP,OMIM,uniprot,ClinVar等数据库中进行检索和过滤,得到各个突变的疾病关联信息。
2.2 直接测序(Sanger法)验证 首先对高通量测序所检测到的可疑致病基因及突变位点行生物信息学分析(参照文献[1]),确定突变的性质;其次对致病性突变进行遗传学分析,并与患儿的临床表型比较,选择临床表型较吻合的突变基因及其致病性突变位点进行验证。本文患儿发现的突变位点,首先利用Sanger直接测序的方法在患儿父母中进行突变位点验证。对新发现的错义突变进行在线 PolyPhen和SFIT预测,并通过多物种蛋白序列比对分析新突变位点的保守性。
2.3 筛选流程 见图2。
2.4 测序结果 4 000种单基因高通量测序和Sanger法验证发现,例1存在NPHS1基因已报道的p.E447K和p.G601A杂合突变,ADCK4基因纯合p.D209H错义突变,为新发现的突变。家系突变分析发现,NPHS1基因p.E447K和p.G601A杂合突变均来自父亲,其哥哥也有相同的基因型,其母亲不携带突变(图3A);患儿父母和哥哥分别携带杂合p.D209H错义突变(图3A)。例2检出COQ6基因的纯合p.R360W错义突变 , 为新发现的突变。
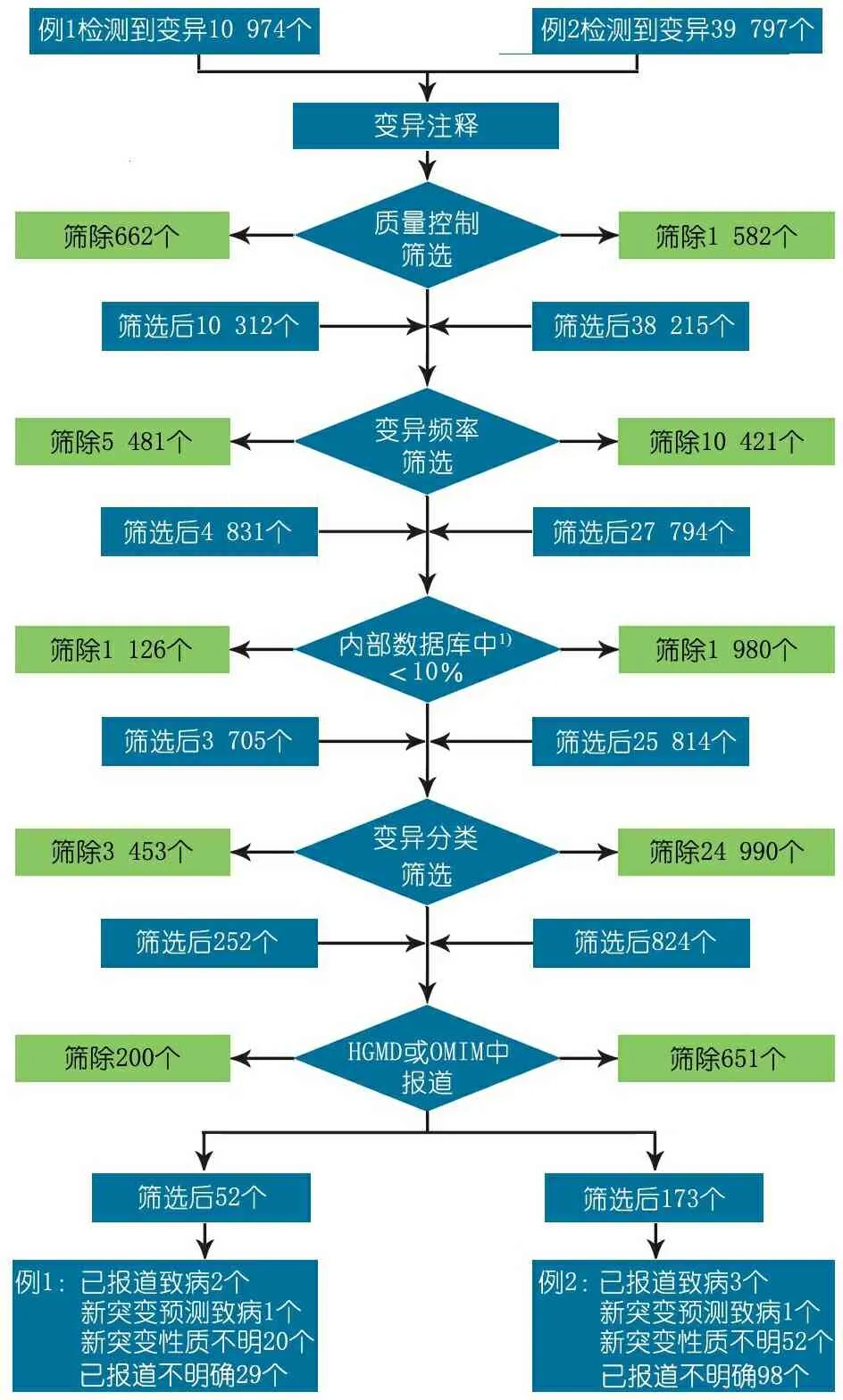
图2 筛选流程图
Fig 2 Filter flow chart
Notes 1)北京德易东方医学转化中心数据库
家系突变分析显示,患儿父母分别携带杂合p.R360W错义突变(图3B)。2例患儿均确诊为线粒体相关肾病。
2.5 测序结果分析ADCK4基因p.D209H突变和COQ6基因的p.R360W突变均为错义突变,经在线软件PolyPhen 和SIFT预测为有害性突变。多物种蛋白序列比对发现,突变位点具有保守性。
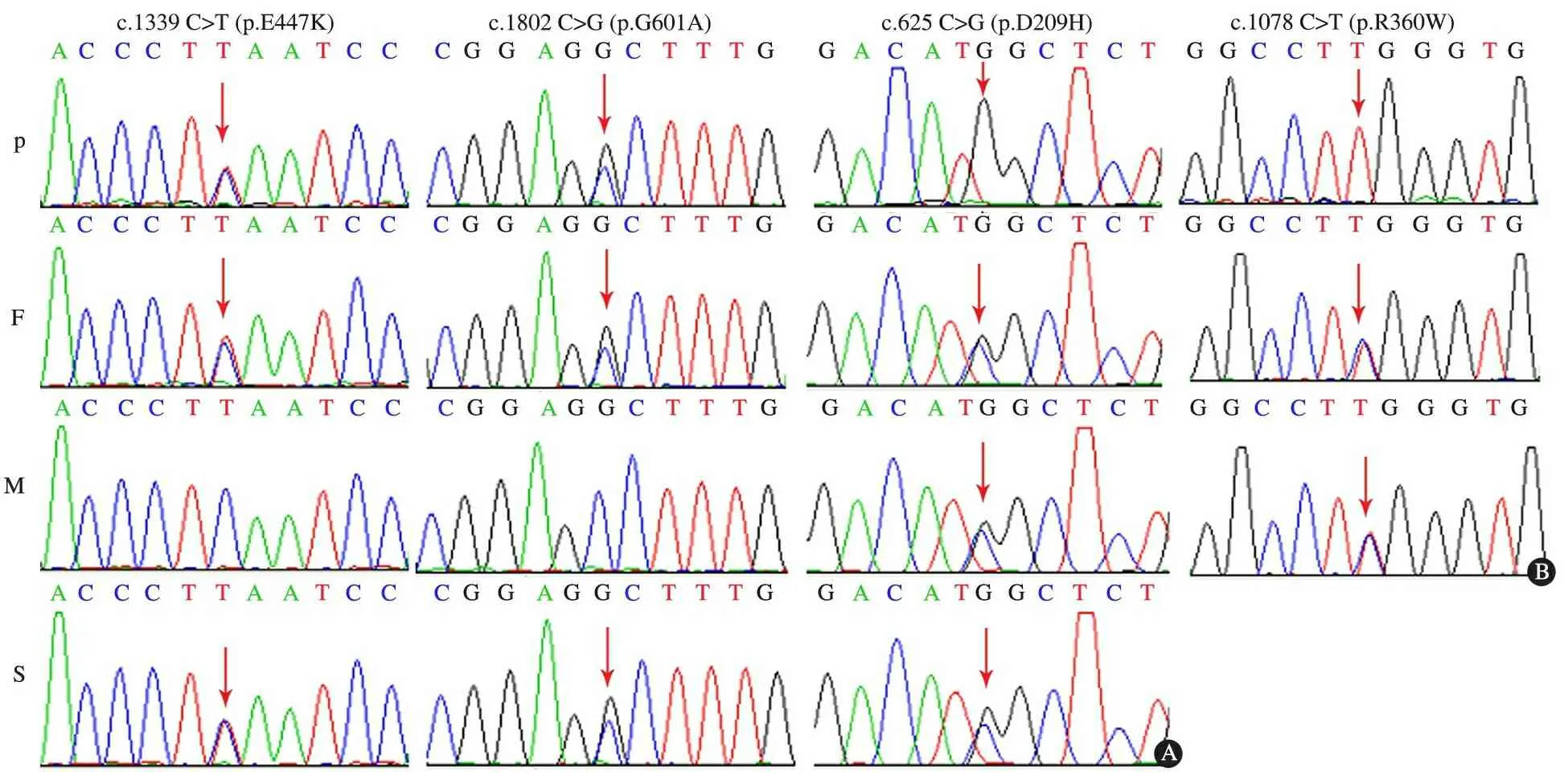
图3 例1和2高通量测序结果
Fig 2 Results of high-flux sequencing in cases 1 and 2
Notes A: mutational analysis inNPHS1 andADCK4 gene in family of patent 1;B: mutational analysis inCOQ6 gene in family of patent 2; P: patient, F: father, M: mother, S: sibling
3 治疗和随访
例1和2基因确诊后均予辅酶Q10 15~30 mg·kg-1·d-1口服,例1随访6个月,尿蛋白+~2+,SCr 80~123 μmol·L-1,BUN 6.0~7.0 mmol·L-1。例2随访6个月,尿蛋白3+~4+,SCr 19~23μmo·L-1,BUN 3.6~4.0 mmol·L-1,临床症状加重不明显。
4 讨论
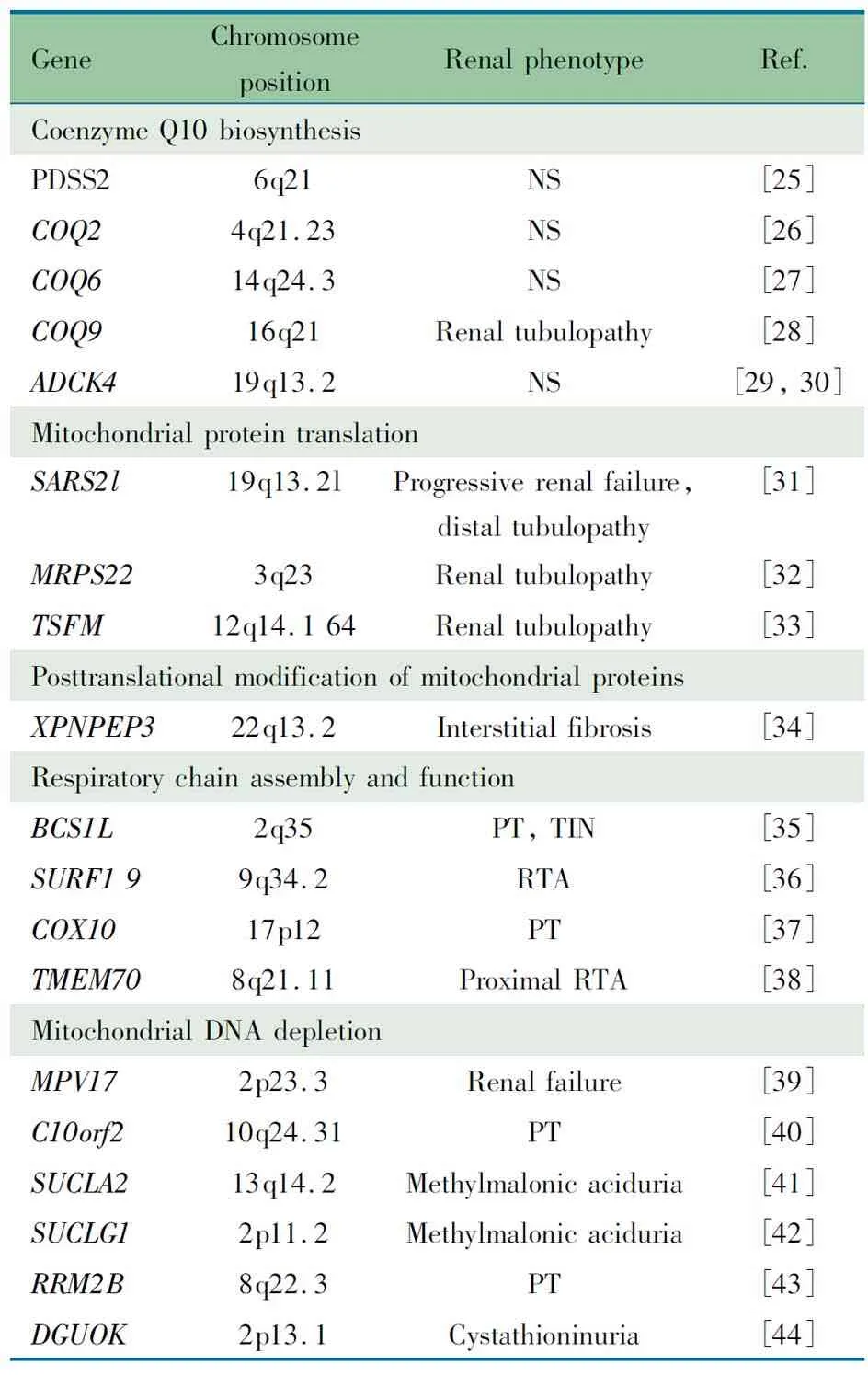
4.1 线粒体基因和核基因突变所致肾病文献复习 线粒体病是最常见和最复杂的遗传性疾病,其遗传方式涉及到线粒体基因(mtDNA)的母系遗传和核基因(nDNA)的孟德尔遗传[2]。各种遗传缺陷导致的线粒体结构和功能异常可引发多个系统能量代谢障碍,常累及脑、骨骼肌、眼、耳、消化、内分泌、心血管及血液系统等[3]。近年来研究发现,多种线粒体基因或核基因遗传缺陷引起的线粒体功能障碍还可累及肾脏(表1,2[4~24]和3[25~44]),称为线粒体相关肾病,临床上对该病常认识不足。线粒体相关肾病可表现为肾小管功能障碍、间质性肾炎、囊性肾病变以及肾小球性病变。线粒体基因和线粒体蛋白编码的核基因突变均可引起肾小球病变,表现为蛋白尿或肾病综合征,既可伴有肾外症状,也可仅表现为肾小球受累,肾脏病理多为FSGS[4,7,16,25~27,29,45~47],对激素治疗及免疫抑制剂治疗无效,其中影响辅酶Q10合成基因突变的一部分患者对辅酶Q10治疗有效[29,48~51]。目前线粒体相关肾病在国内尚未见报道,国外报道也罕见。
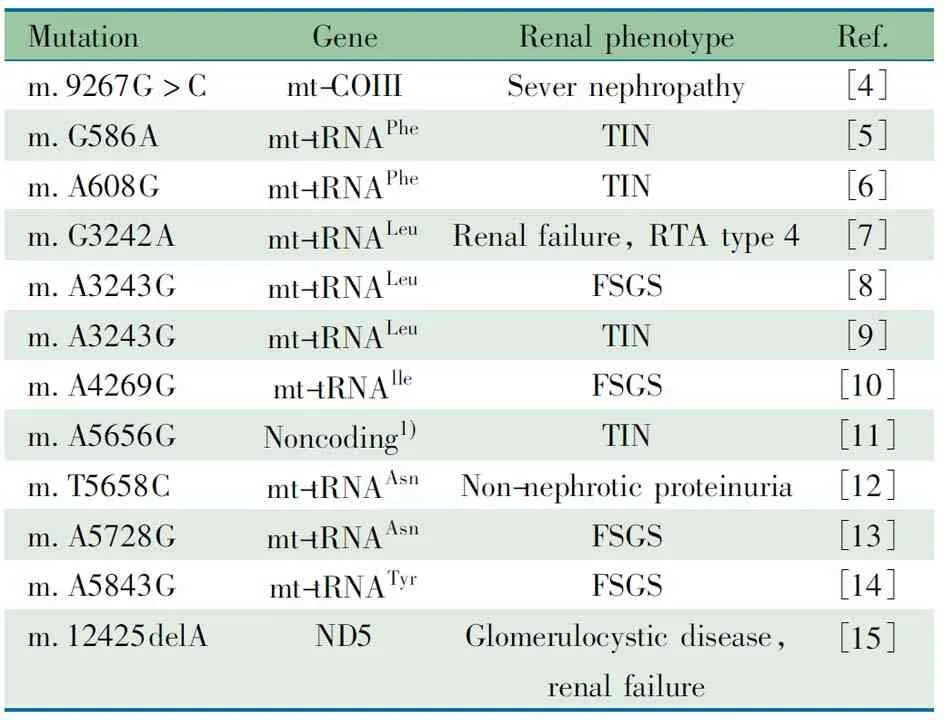
表1 文献报道的线粒体基因点突变相关肾病
Notes 1) Single noncoding base between mt-tRNAAlaand mt-tRNAAsn. mt-tRNA: mitochondrial transfer ribonucleic acid; TIN: tubulointerstitial nephritis; FSGS: focal segmental glomerulosclerosis; RTA, renal tubular acidosis; Phe: phenylalanine; Leu: leucine; Ile: isoleucine; Tyr: tyrosine; Ala: alanine; ND5: NADH dehydrogenase 5
4.2线粒体相关肾病临床表型文献复习本文例1起病年龄为11.7岁,表现为蛋白尿和肾功能受损,无肾外症状,肾脏病理为FSGS。高通量测序和Sanger法验证检出NPHS1基因p.E447K和p.G601A杂合突变,为已报道的致病性突变[52,53],但家系分析发现该2个突变均来自父亲,患儿哥哥也有相同基因型,其母亲不携带这2个突变;因患儿父亲和哥哥均无蛋白尿等表现,推测该2个突变位于相同的等位基因上。例1还检出ADCK4基因p.D209H纯合突变,其父母和同胞分别携带p.D209H杂合突变,经在线软件分析为有害性突变,其突变位点具有保守性,提示该位点应为致病性突变,且为新发现的突变。国外1项研究汇总8个家庭15例ADCK4基因突变患儿,均表现激素耐药肾病综合征,13例有肾活检资料,其中1例为球性硬化,12例为FSGS(包括3例塌陷性FSGS),临床资料完整的9例患儿中,1例有甲状腺肿伴高血压,1例智力发育延迟,1例扩张性心肌病(表4)[29,54]。另一项研究报道了12个家庭26例青少年期起病的患儿,平均发病年龄14.1(10.8~17.0)岁,其中61.5%患儿行肾活检,肾病病理均为FSGS,43.9%有肾病范围蛋白尿,46.1%病例确诊时为慢性肾脏病3~5期,3例偶有惊厥发作,1例智力落后,1例色素性葡萄膜炎[30]。本文患儿仅表现为非肾病水平蛋白尿,尿微量蛋白提示以白蛋白增高为主,肾功能异常,不伴有肾外表型,其临床特点与上述2项研究报道的病例相似,可诊断为线粒体相关肾病,即ADCK4基因相关肾小球疾病。该病可无肾外症状,肾脏表现可以为激素耐药肾病综合征(肾病水平蛋白尿),也可为非肾病水平蛋白尿,易进展为终末期肾病。
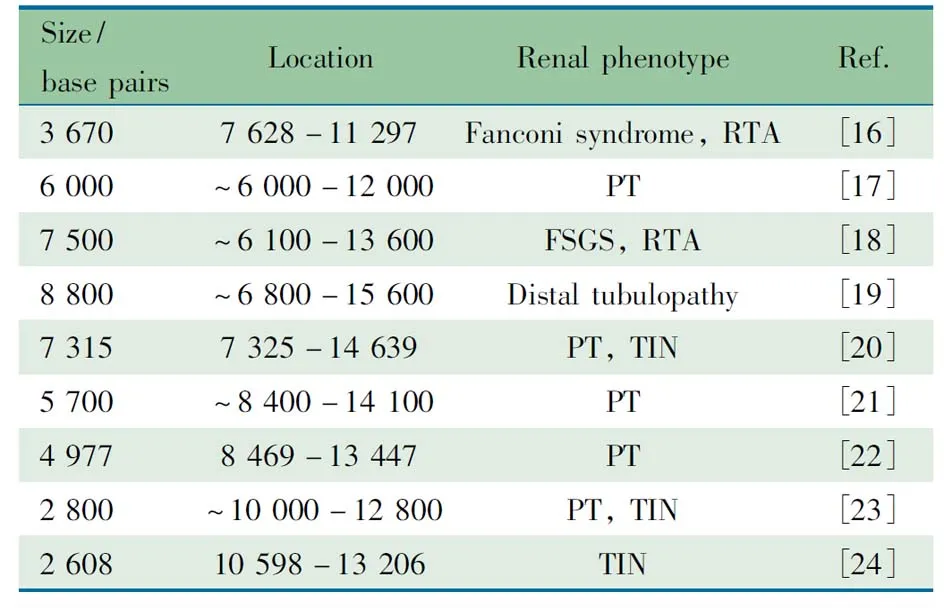
表2 文献报道的线粒体基因缺失相关肾病
Notes TIN: tubulointerstitial nephritis; FSGS: focal segmental glomerulosclerosis; RTA, renal tubular acidosis; PT: proximal tubulopathy
表3 线粒体蛋白编码的核基因相关肾病
Tab 3 Nuclear genes encoding mitochondrial proteins associated nephropathy
GeneChromosomepositionRenalphenotypeRef.CoenzymeQ10biosynthesisPDSS26q21NS[25]COQ24q21.23NS[26]COQ614q24.3NS[27]COQ916q21Renaltubulopathy[28]ADCK419q13.2NS[29,30]MitochondrialproteintranslationSARS2l19q13.2lProgressiverenalfailure,distaltubulopathy[31]MRPS223q23Renaltubulopathy[32]TSFM12q14.164Renaltubulopathy[33]PosttranslationalmodificationofmitochondrialproteinsXPNPEP322q13.2Interstitialfibrosis[34]RespiratorychainassemblyandfunctionBCS1L2q35PT,TIN[35]SURF199q34.2RTA[36]COX1017p12PT[37]TMEM708q21.11ProximalRTA[38]MitochondrialDNAdepletionMPV172p23.3Renalfailure[39]C10orf210q24.31PT[40]SUCLA213q14.2Methylmalonicaciduria[41]SUCLG12p11.2Methylmalonicaciduria[42]RRM2B8q22.3PT[43]DGUOK2p13.1Cystathioninuria[44]
Notes NS: nephrotic syndrome; RTA, renal tubular acidosis; PT: proximal tubulopathy; TIN: tubulointerstitial nephritis
本文例2表现为出生后即起病,多系统受累,累及脑、骨骼肌、心血管和肾脏,符合线粒体病的临床特征。高通量测序发现检出COQ6基因的纯合p.R360W错义突变,经Sanger法验证。家系突变分析显示,患儿父母分别携带杂合p.R360W错义突变。该突变经在线软件分析为有害性突变,且具有保守性,考虑为致病性突变,且为新发现突变。国外1项研究报道了5个家系12例COQ6基因突变患者,均为激素耐药肾病综合征,其中10例伴有感音神经性耳聋,3例为先天性感音神经性耳聋,5例出现感音神经性耳聋年龄较晚(表4),1例伴有惊厥,1例有脑白质异常和惊厥,2例有共济失调和面部畸形[27]。本文例2突出表现为神经、心血管和肾脏等多系统受累,未发现感音神经性耳聋,结合高通量测序结果,应诊断为线粒体相关肾病,即COQ6基因相关肾小球疾病。COQ6基因相关肾小球疾病除肾脏受累外,还可以累及其他系统,最常见的肾外表现为感音神经性耳聋,也可伴有心脏等系统表现。
虽然线粒体相关肾病累及肾小球时肾脏病理光镜下主要是FSGS或塌陷性FSGS,少部分患者为球性硬化(表1、2、3和4),但光镜下肾脏病理改变并非线粒体相关肾病特征性改变[25,27,29,55]。有研究发现,电镜下远端肾小管和集合管细胞中出现颗粒状肿胀的上皮细胞(GSECc)是线粒体相关肾病的特征改变,因IgA肾病、原发性FSGS和间质性肾炎并无GSECc出现[55,56]。但目前文献报道的病例并无该特征性改变。本文例1肾脏病理光镜下显示12肾小球有8个球性硬化,1个为FSGS,免疫荧光显示少量IgG、IgM、IgA、C1q和C3沉积,为非特异性改变 , 电镜下显示足细胞足突部分融合,未观察到GSECc,与国外报道相似。本文例2未行肾活检,肾功能异常与多个肾小球球性硬化严重病变相符,提示疾病进展较快。
COQ6和ADCK4基因突变等引起的线粒体相关肾病对激素和免疫抑制剂治疗无反应,易快速进展为终末期肾病,需要肾脏替代治疗[27,29,49,57]。部分患儿经辅酶Q10治疗后临床症状有所缓解。本文例2未给予激素治疗,例1家长拒绝激素治疗,2例基因诊断明确后均给予大剂量辅酶Q10(15~30 mg·kg-1·d-1)治疗,虽然经治疗临床症状改善不明显,但随访期间临床症状未见加重,而且肾功能监测无明显恶化,这也可能提示线粒体相关肾病(如肾小球球性硬化)可能不可逆。早期发现,早期干预有可能避免不可逆肾损害。
线粒体相关肾病虽然罕见,但该病已经是儿童终末期肾病的重要病因之一[49,57]。提高对该病的认识,早期发现和早期干预该病,有助于儿童慢性肾脏病的防治。
[1]Li GM(李国民),Shen Q,Xu H,et al. Studies on strategy of gene screening in children with nephrotic syndrome. Chin J Evid Based Pediatr(中国循证儿科杂志),2015,10(5):350-356
[2]Lightowlers RN, Taylor RW, Turnbull DM. Mutations causing mitochondrial disease: What is new and what challenges remain?. Science,2015,349(6255):1494-1499
[3]Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet,2005,6(5):389-402
[4]Tabebi M, Mkaouar-Rebai E, Mnif M, et al. A novel mutation MT-COIII m.9267G>C and MT-COI m.5913G>A mutation in mitochondrial genes in a Tunisian family with maternally inherited diabetes and deafness (MIDD) associated with severe nephropathy. Biochem Biophys Res Commun,2015,459(3):353-360
[5]D′Aco KE, Manno M, Clarke C, et al. Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol,2013,28(3):515-519
[6]Tzen CY, Tsai JD, Wu TY, et al. Tubulointerstitial nephritis associated with a novel mitochondrial point mutation. Kidney Int,2001,59(3):846-854
[7]Wortmann SB, Champion MP, van den Heuvel L, et al. Mitochondrial DNA m.3242G > A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction?. Eur J Med Genet,2012,55(10):552-556
[8]Guery B, Choukroun G, Noel LH, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol,2003,14(8):2099-2108
[9]Hirano M, Konishi K, Arata N, et al. Renal complications in a patient with A-to-G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Intern Med,2002,41(2):113-118
[10]Taniike M, Fukushima H, Yanagihara I, et al. Mitochondrial tRNA(Ile) mutation in fatal cardiomyopathy. Biochem Biophys Res Commun,1992,186(1):47-53
[11]Zsurka G, Ormos J, Ivanyi B, et al. Mitochondrial mutation as a probable causative factor in familial progressive tubulointerstitial nephritis. Hum Genet,1997,99(4):484-487
[12]Pinos T, Melia M J, Ortiz N, et al. Identification of the novel mutation m.5658T>C in the mitochondrial tRNA(Asn) gene in a patient with myopathy, bilateral ptosis and ophthalmoparesis. Neuromuscul Disord,2013,23(4):330-336
[13]Meulemans A, De Paepe B, De Bleecker J, et al. Two novel mitochondrial DNA mutations in muscle tissue of a patient with limb-girdle myopathy. Arch Neurol,2007,64(9):1339-1343
[14]Scaglia F, Vogel H, Hawkins EP, et al. Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am J Med Genet A,2003,123A(2):172-178
[15]Alston CL, Morak M, Reid C, et al. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul Disord,2010,20(2):131-135
[16]Liu HM, Tsai LP, Chien YH, et al. A novel 3670-base pair mitochondrial DNA deletion resulting in multi-systemic manifestations in a child. Pediatr Neonatol,2012,53(4):264-268
[17]Campos Y, Garcia-Silva T, Barrionuevo CR, et al. Mitochondrial DNA deletion in a patient with mitochondrial myopathy, lactic acidosis, and stroke-like episodes (MELAS) and Fanconi′s syndrome. Pediatr Neurol,1995,13(1):69-72
[18]Eviatar L, Shanske S, Gauthier B, et al. Kearns-Sayre syndrome presenting as renal tubular acidosis. Neurology,1990,40(11):1761-1763
[19]Goto Y, Itami N, Kajii N, et al. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr,1990,116(6):904-910
[20]Au KM, Lau SC, Mak YF, et al. Mitochondrial DNA deletion in a girl with Fanconi′s syndrome. Pediatr Nephrol,2007,22(1):136-140
[21]Majander A, Suomalainen A, Vettenranta K, et al. Congenital hypoplastic anemia, diabetes, and severe renal tubular dysfunction associated with a mitochondrial DNA deletion. Pediatr Res,1991,30(4):327-330
[22]Mcshane MA, Hammans SR, Sweeney M, et al. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet,1991,48(1):39-42
[23]Szabolcs MJ, Seigle R, Shanske S, et al. Mitochondrial DNA deletion: a cause of chronic tubulointerstitial nephropathy. Kidney Int,1994,45(5):1388-1396
[24]Rotig A, Goutieres F, Niaudet P, et al. Deletion of mitochondrial DNA in patient with chronic tubulointerstitial nephritis. J Pediatr,1995,126(4):597-601
[25]Gasser DL, Winkler CA, Peng M, et al. Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10. Am J Physiol Renal Physiol,2013,305(8):F1228-F1238
[26]Ogaki K, Fujioka S, Heckman MG, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener,2014,9(3):44
[27]Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest,2011,121(5):2013-2024
[28]Duncan AJ, Bitner-Glindzicz M, Meunier B, et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet,2009,84(5):558-566
[29]Ashraf S, Gee HY, Woerner S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest,2013,123(12):5179-5189
[30]Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, et al. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol, 2015
[31]Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet,2011,88(2):193-200
[32]Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet,2007,44(12):784-786
[33]Vedrenne V, Galmiche L, Chretien D, et al. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol,2012,56(1):294-297
[34]O′Toole JF, Liu Y, Davis EE, et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest,2010,120(3):791-802
[35]de Lonlay P, Valnot I, Barrientos A, et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet,2001,29(1):57-60
[36]Tay SK, Sacconi S, Akman HO, et al. Unusual clinical presentations in four cases of Leigh disease, cytochrome C oxidase deficiency, and SURF1 gene mutations. J Child Neurol,2005,20(8):670-674
[37]Valnot I, von Kleist-Retzow JC, Barrientos A, et al. A mutation in the human heme A:farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum Mol Genet,2000,9(8):1245-1249
[38]Honzik T, Tesarova M, Mayr JA, et al. Mitochondrial encephalocardio-myopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child,2010,95(4):296-301
[39]Uusimaa J, Evans J, Smith C, et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur J Hum Genet,2014,22(2):184-191
[40]Prasad C, Melancon SB, Rupar CA, et al. Exome sequencing reveals a homozygous mutation in TWINKLE as the cause of multisystemic failure including renal tubulopathy in three siblings. Mol Genet Metab,2013,108(3):190-194
[41]Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain,2007,130(Pt 3):853-861
[42]Ostergaard E, Schwartz M, Batbayli M, et al. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr,2010,169(2):201-205
[43]Bourdon A, Minai L, Serre V, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet,2007,39(6):776-780
[44]Tadiboyina VT, Rupar A, Atkison P, et al. Novel mutation in DGUOK in hepatocerebral mitochondrial DNA depletion syndrome associated with cystathioninuria. Am J Med Genet A,2005,135(3):289-291
[45]Alston CL, Morak M, Reid C, et al. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul Disord,2010,20(2):131-135
[46]Jackson CB, Bauer MF, Schaller A, et al. A novel mutation in BCS1L associated with deafness, tubulopathy, growth retardation and microcephaly. Eur J Pediatr,2015
[47]Klootwijk ED, Reichold M, Helip-Wooley A, et al. Mistargeting of peroxisomal EHHADH and inherited renal Fanconi′s syndrome. N Engl J Med,2014,370(2):129-138
[48]Doimo M, Trevisson E, Airik R, et al. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim Biophys Acta,2014,1842(1):1-6
[49]D′Aco KE, Manno M, Clarke C, et al. Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol,2013,28(3):515-519
[50]Carney EF. Tubular disease: mistargeted protein disrupts mitochondrial metabolism in inherited Fanconi syndrome. Nat Rev Nephrol,2014,10(3):125
[51]Lee IC, Lee NC, Lu JJ, et al. Mitochondrial depletion causes neonatal-onset leigh syndrome, myopathy, and renal tubulopathy. J Child Neurol,2013,28(3):404-408
[52]Aya K, Tanaka H, Seino Y. Novel mutation in the nephrin gene of a Japanese patient with congenital nephrotic syndrome of the Finnish type. Kidney Int,2000,57(2):401-404
[53]Beltcheva O, Martin P, Lenkkeri U, et al. Mutation spectrum in the nephrin gene (NPHS1) in congenital nephrotic syndrome. Hum Mutat,2001,17(5):368-373
[54]Malaga-Dieguez L, Susztak K. ADCK4 "reenergizes" nephrotic syndrome. J Clin Invest,2013,123(12):4996-4999
[55]Kobayashi A, Goto Y, Nagata M, et al. Granular swollen epithelial cells: a histologic and diagnostic marker for mitochondrial nephropathy. Am J Surg Pathol,2010,34(2):262-270
[56]Imasawa T, Tanaka M, Yamaguchi Y, et al. 7501 T > A mitochondrial DNA variant in a patient with glomerulosclerosis. Ren Fail,2014,36(9):1461-1465
[57]Rahman S, Hall AM. Mitochondrial disease--an important cause of end-stage renal failure. Pediatr Nephrol,2013,28(3):357-361
(本文编辑:丁俊杰)
Mitochondrial nephropathy in two children and literature review
LIGuo-min1,5,SUNLi1,5,SHENQian1,XUHong1,FANGXiao-yan1,CAOQi1,LIUHai-mei1,ZHAIYi-hui1,WUBing-bing2,LIUXue-guang3,YANGQing4
(1DepartmentofNephrologyandRheumatology,Children′sHospitalofFudanUniversity,Shanghai201102; 2MedicalTranslationalCenterofChildren′sHospitalofFudanUniversity,Shanghai201102; 3DepartmentofPathology,ShanghaiMedicalCollegeofFudanUniversity,Shanghai200023; 4The2ndAffiliatedHospitalandYuyingChildren′sHospitalofWenzhouMedicalUniversity,Wenzhou325027,China; 5hasequalcontribution)
XU Hong,E-mail:hxu@shmu.edu.cn
ObjectiveTo summarize and review the clinical data of two children with mitochondrial nephropathy so as to improve it′s knowledge. MethodsClinical data of two cases with mitochondrial nephropathy were summarized, including clinical manifestations, laboratory findings, renal pathological changes and family investigation. This study used next generation sequencing to screen 4 000 genes, including the 40 genes known to be associated with mitochondrial disease. Significant variants detected by next generation sequencing were confirmed by conventional Sanger sequencing and segregation analysis was performed using parental DNA samples.ResultsIn two cases, one is a boy, the other is a girl. Age onset of the girl was 11.7 years. She presented with proteinuria, renal dysfunction, no extrarenal symptoms and focal segmental glomerulosclerosis (FSGS) in renal biopsy. Heterozygous p.E447 and p.G601A mutations inNPHS1 and homozygous p.D209H mutation inADCK4 gene were detected and confirmed by next-generation sequencing and conventional Sanger sequencing, respectively. Family analysis showed that the girl had same genotype inNPHS1 gene with her father and sibling, and her homozygous p.D209H mutation inADCK4 gene was from parents. The boy presented with congenital heart disease and mental retardation after birth, and nephrotic syndrome in few months later. Homozygous p.R360W mutation inCOQ6 gene was identified and confirmed by next-generation sequencing and Sanger sequencing, respectively. Family analysis showed that homozygous p.R360W mutation in COQ6 gene inherited from his parents. Missense p.D209H and p.R360W mutations were damaging by prediction online PolyPhen and SIFT software. Protein multiple alignment showed site in p.D209H and p.R360W mutations both were conservative.ConclusionTwo cases with renal phenotype were caused by casual mutations inADCK4 andCOQ6 gene, respectively. These two cases could be diagnosed as mitochondrial nephropathy. One case with mutation inADCK4 gene presented with proteinuria, renal dysfunction, no extrarenal symptoms and FSGS in renal biopsy. The other with mutation inCOQ6 gene had nephrotic syndrome, except congenital heart disease and mental retardation.
Mitochondrial disease; Proteinuria; Nephrotic syndrome;ADCK4 gene;COQ6 gene
复旦大学附属儿科医院人才工程-学科带头人(1125)培育计划
1 复旦大学附属儿科医院肾脏风湿科 上海,201102;2 复旦大学附属儿科医院医学转化中心 上海,201102; 3 复旦大学上海医学院病理教研室 上海,200023;4 温州医科大学附属第二医院育英儿童医院 温州,325027;5 共同第一作者
徐虹,E-mail:hxu@shmu.edu.cn
10.3969/j.issn.1673-5501.2015.06.006
2015-09-30
2015-11-27)
