液液萃取-HPLC-ICPMS联用技术测定水体中甲基汞
2015-04-26周慧兰
陈 贺,周慧兰
成都市环境监测中心站,四川 成都 610072
甲基汞是一种具有生物高毒性和易生成性的物质。目前水体中甲基汞的检测方法主要有气相色谱法、原子荧光法和电感耦合等离子体质谱法。由于蒸馏-乙基化-吹扫捕集气相色谱蒸汽发生原子荧光(CVAFS)联用法[1-2]步骤繁琐,巯基棉富集-气相色谱法(GC)法[3-4]重现性差,选择性差,且萃取试剂毒性大,所以最近几年液相色谱-原子荧光联用法在甲基汞的检测方面得到了深入的研究[5-7]。与液相色谱-原子荧光联用法相比,液相色谱-电感耦合等离子体质谱法(LC-ICP/MS)在准确度和灵敏度等性能上有更大的优势,但是LC-ICP/MS的仪器灵敏度[8-9]不能满足《地表水环境质量标准》中甲基汞的标准限值(1 ng/L)的要求,所以选择合适的前处理富集方法是降低甲基汞测定检出限的有效解决方法。由于半胱氨酸-乙酸铵体系的毒性较小,以该体系作为流动相和萃取液成为近年来甲基汞分析方法的研究热点,杨坪等[6-7]开发了二氯甲烷萃取、半胱氨酸-乙酸铵反萃取、液相色谱-原子荧光联用法测定甲基汞的方法。该文拟在上述二氯甲烷萃取、半胱氨酸-乙酸铵反萃取前处理方法的基础上,采用液相色谱-电感耦合等离子体质谱法作为分析仪器,探索建立一种灵敏度高、选择性好的甲基汞测定方法,能够满足《地表水环境质量标准》中甲基汞限值要求的分析方法。
1 实验部分
1.1 仪器和试剂
1200型液相色谱,7700X型电感耦合等离子体质谱。
色谱条件:色谱柱(zorbax SB-C18,4.6 mm×250 mm ×5 μm);流动相:10 mmol/L乙酸铵,0.12%半胱氨酸,氨水调pH至7.5,8%甲醇水溶液;流速为1.0 mL/min;进样环体积为100 μL。
ICP-MS参数:入射功率1 550 W,采样深度810 mm,载气流速约0.16 L/min,No gas模式,同位素为202Hg,积分时间为0.5 s。
1.2 主要试剂和材料
甲醇,HPLC级,DikmaPure;二氯甲烷,HPLC级,DikmaPure;半胱氨酸,Sigma;乙酸铵,HPLC级,CNW;氨水,HPLC级,CNW;氯化钠,优级纯,科龙试剂,400℃烘烤4 h;甲基汞标准溶液,(60.2±2.3)μg/g,9℃时相当于 48.2 mg/L,中国计量科学院;Mili-Q纯水。
2 结果与讨论
2.1 前处理
实验条件下采用2.41 ng/L的空白加标样进行前处理条件的优化实验。
2.1.1 水样酸碱度和盐度
考察了酸性、中性、碱性水样的回收率情况,实验结果表明,中性条件的水样回收率最好,酸性和碱性水样的回收率较差,如图1所示。通过在水样中添加不同量的氯化钠(NaCl)考察水样盐度对回收率的影响,结果表明,盐度对回收率影响很小(图2),因为添加NaCl可以减轻实际水样萃取时的乳化现象,所以本文选择NaCl添加量为10 g。

图1 水样酸碱度对回收率的影响

图2 水样NaCl添加量对回收率的影响
2.1.2 二氯甲烷萃取条件
优化了二氯甲烷的萃取次数和萃取时间,由于反萃取使用的是50 mL比色管,体积较小,所以二氯甲烷萃取次数的优化实验第1次、第2次、第3次二氯甲烷用量分别为10、20、30 mL。从图3可见,萃取2次即可达到稳定的回收率。萃取时间对回收率的影响见图4,可以看出,每次萃取10 min可以达到平衡。
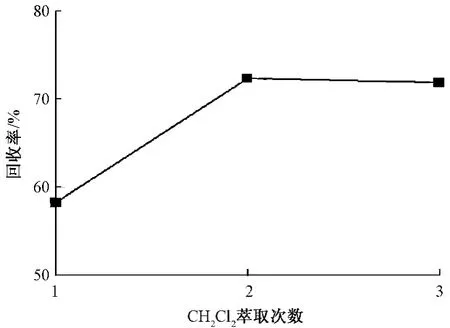
图3 二氯甲烷萃取次数对回收率的影响
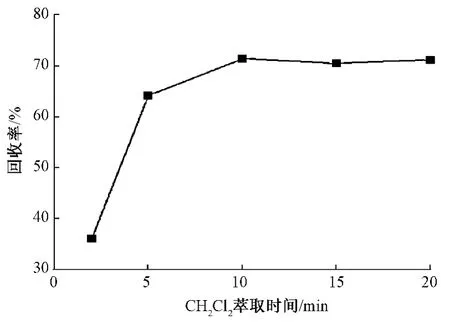
图4 二氯甲烷萃取时间对回收率的影响
2.1.3 反萃取条件
由图5可知,反萃取液用量越大,回收效果越好,但是反萃取液的增加会增大检出限。由图6可知,随着反萃取时间的延长,回收率提高,20 min后回收率下降,分析原因可能是由于反萃取过程需要不断打开比色管塞放气体,造成甲基汞损失。综合考虑,选择反萃取液用量为5 mL,反萃取时间20 min。

图5 反萃取液用量对回收率的影响
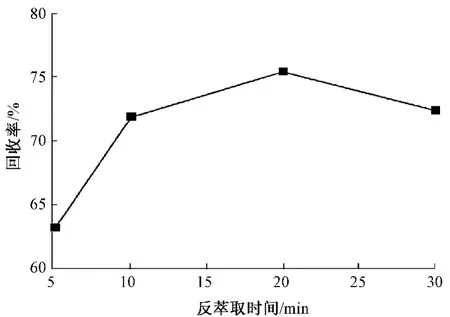
图6 反萃取时间对回收率的影响
优化后的前处理过程:取均匀水样1 L,置于1 L分液漏斗中,加约10 g氯化钠,用30 mL二氯甲烷萃取10 min后,用20 mL二氯甲烷萃取10 min。萃取液收集至50 mL比色管中,加5 mL反萃取溶液(1%半胱氨酸-0.8%乙酸铵溶液)进行反萃取,振荡20 min后,吸取水层溶液进样。
2.2 性能指标
2.2.1 分离图
甲基汞(0.494 μg/L)和乙基汞(0.494 μg/L)的色谱分离图见图7。
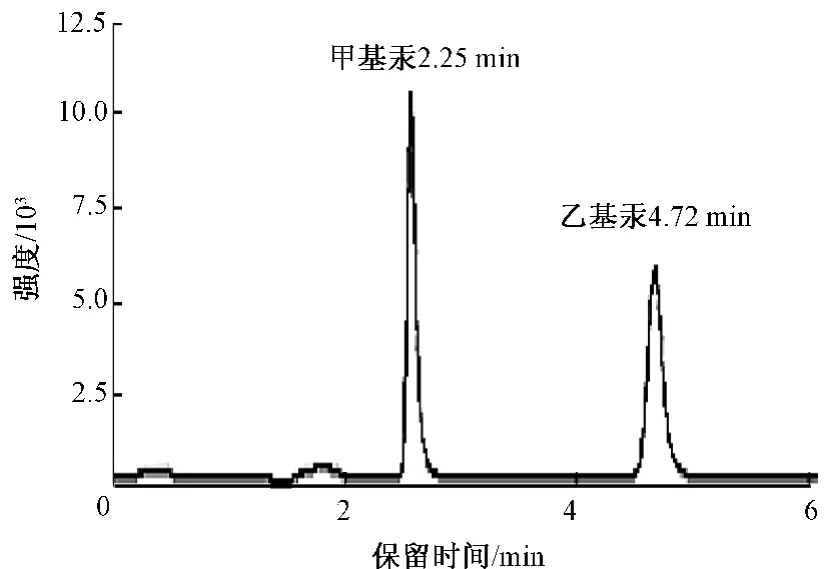
图7 色谱分离图
2.2.2 线性、检出限、精密度
按照前处理方法和色谱质谱条件,完成分析性能参数测定。曲线为外标法,用反萃取液配制质量浓度分别为 0、0.096 3、0.482、1.93、4.82 μg/L 标准溶液系列,实验结果表明线性较好,线性相关系数为0.999 7。按照HJ 168—2010的规定,配制质量浓度为0.482 ng/L的空白加标样7份进行全程序分析,检出限MDL=3.143×S,式中S为7次平行实验结果的标准偏差,计算得方法检出限为0.1 ng/L,低于《地表水环境质量标准》(GB 3838—2002)的限值(1 ng/L)。配制高、中、低3种质量浓度(9.65、2.41、0.482 ng/L)的空白加标样各6份,分别进行全程序分析,考察精密度和准确度,精密度(相对标准偏差RSD)为6.6% ~13%,准确度(相对误差)为-21.6% ~-44.4%。
2.2.3 实际水样的测定
通过分别测试高、中、低3种浓度的地表水、生活污水、工业废水加标样的回收率(各平行测定3份水样),对实际样品加标回收率进行了考察,结果如表1所示。
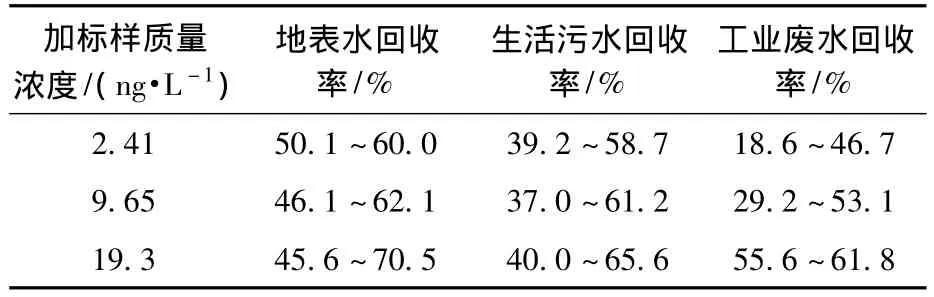
表1 实际水样回收率
由表1可知,该方法的回收率较稳定,适于实际样品中甲基汞的测定。实际样品的回收率不高,且基体越复杂,回收率的相对偏差越大,主要是由于基体中悬浮物和采样、萃取容器对甲基汞有一定的吸附作用,基体中物质对甲基汞萃取有干扰。
2.3 与其他方法的比较
目前水体中甲基汞的测定方法主要有表2中的5种方法。与气相色谱法相比,HPLC-ICP/MS法和HPLC-AFS法是更适用于监测甲基汞的分析方法。该方法与无富集过程的HPLC-ICP/MS[8-9]相比,检出限低2个数量级,完全满足《地表水环境质量标准》(GB 3838—2002)中甲基汞限值的要求;与 HPLC-AFS法[6-7]相比,进样量少,峰形好,检出限更低,并具有更好的选择性。

表2 水体中甲基汞分析方法比较
2.4 讨论
无机汞(Hg2+和元素汞)的干扰会引起本方法的灵敏度降低,主要表现在2个方面:一是体系中无机汞浓度高,会引起基线噪声的升高,致使甲基汞的检出限大幅提高;二是由于Hg2+和甲基汞保留时间相差较小,Hg2+峰的拖尾现象会影响甲基汞的测定,如图8所示。
元素汞的干扰主要是富集在管路中引起大的基线噪声,通过冲洗管路可消除干扰,Hg2+主要存在于试剂和样品中。该文研究了Hg2+浓度对甲基汞准确定性、定量的影响,实验结果表明,为保证其能满足GB 3838—2002中甲基汞限值(1.0 ng/L)的要求,体系中Hg2+的质量浓度不能大于0.05 μg/L,以保证有足够低的基线噪声;浓缩后的样品甲基汞质量浓度为0.1 μg/L时,质量浓度大于1 μg/L的无机汞对甲基汞定量的准确度有影响,约50 μg/L的无机汞的峰拖尾会覆盖甲基汞的出峰,影响准确定性。
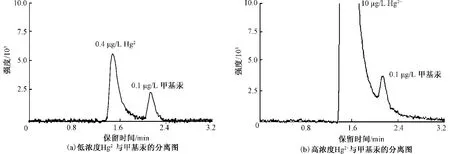
图8 Hg2+对甲基汞的影响
为了减少无机汞污染,可采取的措施:所用试剂和药品需进行空白值实验,防止无机汞含量过高引起基线噪声过大;当基线噪声过高是由LC管路污染引起时,取下色谱柱,用高浓度的半胱氨酸和乙酸铵的水溶液冲洗;实验中所用的器皿需用10%HNO3浸泡24 h,以去除无机汞,不宜用硝酸浸泡的器皿可用半胱氨酸和乙酸铵溶液反复清洗后,用去离子水清洗。
3 结论
采用二氯甲烷萃取,半胱氨酸-乙酸铵溶液反萃取作为前处理方法,与液相色谱-电感耦合等离子体质谱法联用,开发了一种灵敏测定水中甲基汞的方法。该方法具有检出限低、准确度高、回收率稳定和干扰小的优点。无机汞的污染是影响方法检出限的重要因素,可通过采用高纯试剂、半胱氨酸-乙酸铵溶液清洗管路和进样针以及硝酸溶液浸泡器皿等方法消除。
[1]EPA Method 1630 Methylmercuryin waterby distillation,aqueous ethylation,purge and trap,and CVAFS[S].
[2]蒋红梅,冯新斌,梁琏,等.蒸馏-乙基化GC-CVAFS法测定天然水体中的甲基汞[J].中国环境科学,2004,24(5):568-571.
[3]祁辉,刘爱民,黄业茹,等.巯基棉富集-毛细柱气相色谱法测定环境水中的甲基汞[J].中国环境监测,2010,26(4):33-36.
[4]刘保献,李新中,常淼,等.固相萃取 气相色谱法测定饮用水中甲基汞[J].环境监测管理与技术,2011,23(1):54-56,60.
[5]尚晓虹,赵云峰,张磊,等.水产品中甲基汞测定的液相色谱-原子荧光光谱联用方法的改进[J].色谱,2011,29(7):667-672.
[6]杨坪,钱蜀.环境样品分析新方法及其应用[M].北京:科学出版社,2010:110-113.
[7]陈邵鹏,顾海东,秦宏兵.高效液相色谱原氢化物发生原原子荧光光谱联用技术测定水中烷基汞[J].中国环境监测,2012,28(5):79-82.
[8]张兰,陈玉红,施燕支,等.高效液相色谱-电感耦合等离子体质谱联用技术测定二价汞、甲基汞、乙基汞与苯基汞[J].安捷伦环境科学专栏,2009,28(5):772-775.
[9]王征,游飞明,邱秀玉,等.HPLC-ICP-MS法测定水样中的甲基汞、乙基汞和无机汞[J].福建分析测试,2009,18(1):28-31.
