胞嘧啶的合成工艺改进
2015-04-23郭琳娜李欣倩毛武涛
郭琳娜,李欣倩,毛武涛
(南阳师范学院化学与制药工程学院,河南南阳 473061)
胞嘧啶(7)是生物体中参与DNA和RNA合成的重要活性原料,也是精细化工、农药和医药的重要中间体,在化学化工和药物化学领域具有广泛应用前景[1-3]。
目前,7的合成主要采用官能团转化法和Pinner合成法[4-5]。这些方法均以 3-乙氧基丙烯腈(Ⅰ),3,3-二乙氧基丙烯腈(Ⅱ)或者其混合物为原料。而制备Ⅰ和Ⅱ均需通过中间体——3-羟基丙烯腈金属盐(Ⅲ)。而Ⅲ的制备由乙腈、一氧化碳、氯乙烷和乙醇钠等在高温高压下反应。以上合成7的方法均存在原料制备困难,操作复杂、危险性大和污染较严重等不足。
本文在文献方法的基础上,优化了一条合成7的路线。以氰乙酸乙酯(1)、尿素(2)和原甲酸三乙酯(3)为原料,经缩合反应制得2-氰基-3-脲基丙烯酸乙酯(4);4在强碱性条件下合环得5-乙氧羰基胞嘧啶(5);5经水解和脱羧合成了7(Scheme 1),总收率65.7%,其结构经1H NMR和ESI-MS确证。
在4的合成中,考察了原料比[r=n(2)∶n(3)∶n(1)]对其收率的影响。结果发现,在较佳反应条件[1 2.0 mol,r=1.5 ∶1.5 ∶1.0,回流反应4 h]下,4 的收率从 69.0%[7]提高至89.1%。
环合反应是合成7的关键步骤,本文对其工艺进行了优化,得出异丙醇/异丙醇钠的最佳催化体系,使5的收率从41%[7]提高至92%。
在脱羧反应中,催化剂对其影响较大。本文采用氯化铵/氯化亚铜协同催化,缩短了反应时间,且收率从文献[6]的 60%提高至 87.0%。同时,采用缓缦加入乙醚的方法加速了7的结晶析出,且纯度较高,蒸馏回收的乙醚可重复使用,节约成本。
改进工艺具有原料易得,工艺合理简便,反应条件温和,操作简单,环境友好等优点;7的纯度高,且总收率达 65.7%,较文献[6]方法有较大幅度地提高。
1 实验部分
1.1 仪器与试剂
Bruke-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Trace DSQ FINNIGSN型质谱仪。
1~3,工业品;其余所用试剂均为分析纯。
1.2 合成
(1)4的合成
在圆底烧瓶中依次加入2 180 g(3.0 mol),3 384 g(3.0 mol)和 1 226 g(2.0 mol),搅拌下回流反应4 h。静置冷却,抽滤,滤饼用丙酮洗涤,干燥得白色固体 4 327.5 g,收率 89%,m.p.213℃ ~215℃(215 ℃[7]);1H NMR(CDCl3)δ:1.35(t,J=7.2 Hz,3H,CH3),4.25(q,J=7.2 Hz,2H,OCH2),7.65(br s,1H,NH),7.83(br s,1H,NH),8.19(s,1H,CH),11.5(br s,1H,NH)。
(2)5的合成
在圆底烧瓶中加入异丙醇150 mL,搅拌下分批加入金属钠3.45 g(0.15 mol),待金属钠完全溶解后再加入4 18.3 g(0.1 mol),回流反应3 h。冷却至室温,抽滤,滤液为A溶液。滤饼倒入烧杯中,边搅拌边加水直至固体全部溶解。过滤,滤液用稀盐酸调至pH 6~7,有沉淀生成;抽滤,滤饼用少量丙酮洗涤得滤饼B。将溶液A静置析晶,抽滤,得滤饼C。合并滤饼B和滤饼C,真空干燥得白色固体5 9.5 g,收率52%,m.p.268 ℃ ~280℃(分解)(272 ℃ ~ 282 ℃[8]);1H NMR δ:1.29(t,J=7.6 Hz,3H,CH3),4.24(q,J=7.6 Hz,2H,OCH2),7.63(br s,1H,NH),8.26(s,1H,CH);ESI-MS m/z:Calcd for C7H9N3O3{[2M+H]+}367.0,found 366.8;{[M+H]+}184.0,found 184.1。
(3)6的合成

Scheme 1

表1 r对4收率的影响*Table 1 Effect of r on yield of 4
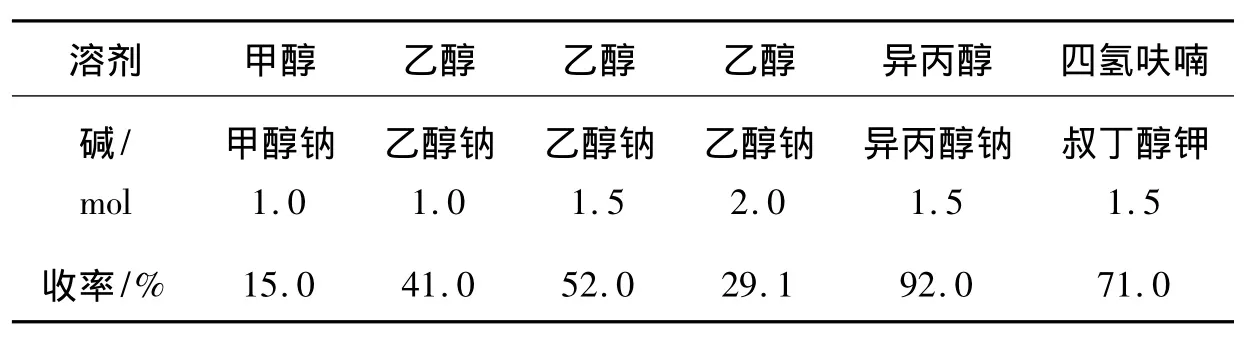
表2 碱及其用量对5收率的影响*Table 2 Effect of alkali and amount on yield of 5
在圆底烧瓶中加入10%NaOH溶液200 mL和 5 18.3 g(0.1 mol),搅拌下回流反应 15 min。冷却至室温,用6%盐酸酸化至pH 4,有白色沉淀生成,抽滤,滤饼用水洗涤,真空干燥得白色固体6 14.3 g,收率92.3%,m.p.258 ℃ ~259 ℃(256℃ ~ 257 ℃[8]);1H NMR δ:7.76(br s,1H,NH),7.89(br s,1H,NH),8.87(s,1H,CH),10.71(br,s,1H,CO2H)。
(4)7的合成
在圆底烧瓶中加入6 7.75 g(50 mmol),氯化铵2.8 g,氯化亚铜 0.2 g 及喹啉 60 mL,氮气保护,搅拌下回流反应至有较少的气泡移除。静置过夜,有少量固体析出,小心加入乙醚50 mL,使反应所得固体从喹啉中析出。过滤,滤饼用乙醚洗涤,干燥后用冰乙酸结晶得白色晶体7 4.83 g,收率 87.0%,m.p.>280 ℃;1H NMR δ:5.90(d,J=6.6 Hz,1H,CH),7.61(d,J=6.6 Hz,1H,CH),8.54(br s,1H,NH),8.60(br s,1H,NH);ESI-MS m/z:Calcd for C4H5N3O{[2M+H]+}223.0,found 223.0;{[M+H]+}112.0,found 112.1。
2 结果与讨论
2.1 4 的合成
实验中发现,在合成4时,原料比r[n(2)∶n(3)∶n(1)]对收率有较大影响(表1)。由表1可见,当 r=1.0 ∶1.0 ∶1.0 时,收率较低(62.5%);当 r=1.5 ∶1.5 ∶1.0 时,收率有大幅提高;当 r=2.0 ∶1.5 ∶1.0 时,收率虽有提高,但幅度不大。可见2的用量对反应有重要影响。
从成本等综合因素考虑,最佳r=1.5∶1.5∶1.0,收率从文献[7]方法的 69.0%提高至 89.1%。
2.2 5 的合成
实验发现,在5的合成中,碱的类型及其用量均对5的收率有较大的影响(表2)。从表2可见,碱性强弱对收率具有显著影响,除叔丁醇钾外,在其它因素不变的情况下,一般碱性增强收率具有明显增加,特别是以异丙醇钠为碱,在异丙醇体系中收率最高(92%)。在已有的文献报道中,在该步反应中所用溶剂均为甲醇或乙醇6-7],碱的用量也各有不同[6-7]。本文通过筛选试剂并通过改变试剂,大幅度地提高了收率。
2.3 7 的合成
胞嘧啶环上羧基的脱除,需要借助催化剂实现。一般可用的催化剂包括氯化铵、固体硫酸-硫酸铝铁、氯化铜、氯化亚铜、硫酸铜。在已有的报道中,一般均使用单一催化剂[6]。本文研究则采用氯化铵、氯化亚铜协同催化,其效果较好,反应时间短,收率较高(87.0%)。
本实验另一个突出的特点在于溶剂的选用对产品的结晶和析出具有显著影响。一般情况在喹啉易于反应脱除羧基,但是产品不易结晶析出,为了使7易于分离,本文使用乙醚便可析出纯度较高的成品,同时提高了7的纯度,且乙醚蒸馏后可重复使用,节约成本。
[1] 扈田进,龚爱琴,何秋.地西他滨的合成[J].广东化工,2013,40(15):47 -48.
[2]王相承,袁天平,李建.一锅法合成N4-乙酰-5-氟胞苷的新工艺研究[J].应用化工,2011,40(3):486 -488.
[3]王亚丽,王振华,曾宪垠.疫苗佐剂胞嘧啶-鸟嘌呤寡脱氧核苷酸的研究进展[J].中国畜牧兽医,2011,38(6):49 -51.
[4]蔡东,商琳琳,贾云宏,等.胞嘧啶的合成研究进展[J].化工中间体,2009,(2):14 -17.
[5] 霍利春,李春新,刘茵.胞嘧啶合成新工艺[J].甘肃石油和化工,2012,(2):28 -32.
[6]Oogami S M.Preparation of cytosine-5-carboxylic acid[P].JP 58 185 569,1983.
[7]Calvert W W.The reactions of orthoesters with ureas.A new synthesis of pyrimidines[J].J Am Chem,1953,75:671-675.
[8]Ulbricht T L V,Price C C.The synthesis of some pyrimidine metabolite analogs[J].J Org Chem,1956,21:567-571.
