全外显子组测序:儿童疾病诊断新思路
2015-04-21马丁艾遥马丁艾路王慧君周文浩
马丁艾遥 马丁艾路 王慧君 周文浩
·综述·
全外显子组测序:儿童疾病诊断新思路
马丁艾遥1,4马丁艾路3,4王慧君2周文浩2
纵观人类遗传学的发展,不断有新兴技术被运用于疾病病因的研究中。从各种流行病学研究设计到如今广泛采用的以假说为导向的候选基因法[1],从以家庭为基础的遗传连锁研究[2~4]到全基因组关联研究(GWAS)[5~8],虽然这些方法均能在不同程度上反映疾病的遗传易感性,但仍不可避免地存在许多限制[9~13]。而人类基因组图谱的完成和新一代高通量测序技术的出现,为疾病病因的研究提供了全新的思路[14]。外显子组序列仅占全基因组序列的1%,但大多数与疾病相关的变异位于外显子区[15~17],基因组定向捕获工具的出现使外显子组测序成为可能。近5年在PubMed上发表的外显子组研究相关文章已达2 000余篇,对数百种疾病展开了深入研究,临床诊断的文章也开始报道,可见利用全基因组外显子测序(WES)寻找人类疾病的致病基因,是近年发展起来的研究疾病致病机制的重要方法之一。
1 WES应用于临床诊断的基本策略
WES是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。大致分为3个步骤:①将人类基因组中所有已知的编码蛋白质的基因序列进行富集,即对全基因组外显子进行捕获、扩增;富集后,待测序序列总量大幅降低,但基因信息总量却下降不多。②应用新一代测序技术对富集保留的DNA序列进行高覆盖深度的测序,高质量、高通量地获取所有基因的编码蛋白质区域的碱基突变信息。③通过生物信息学分析及验证,找到致病基因[18]。WES相对于基因组重测序成本较低,对研究已知基因的单核苷酸多态性(SNP)、剪接插入缺失(Indel) 等具有较大的优势。
以单基因疾病研究方案为例,首先需要按照疾病表型对家系成员进行严格筛查,明确其患病情况。再找出该疾病已有的研究背景和相关致病基因报道,通过传统PCR测序方法对已知的疾病基因相关变异进行验证和初筛;确认所研究的样本中未发现相关的基因变异,挑选一个或数个相同疾病家系的核心成员进行WES。每个家系中的患病个体选取3~5个样本,正常个体选取1~2名作为对照进行研究。按照疾病模型(常染色体显性遗传、常染色体隐性遗传等)及样品的家系信息对测序得到的结果进行分析,缩小候选变异的范围,经过多种注释、筛选后过滤对功能无影响的变异及公共数据库中的常见变异,再使用传统PCR测序进行样本扩大化验证及相关的功能研究,最终确定疾病相关变异。
若为散发样本,由于样本间没有血缘关系,遗传背景相差较大,测序得到的结果也较难分析。使用散发样本进行WES时需要更大的样本量(>30例),方能得到更为准确和有价值的结果。对大量患病个体的测序数据进行多样本分析,从而确定候选疾病相关变异,再用传统PCR测序在其他的相同疾病患病个体和正常人群中做进一步验证。
2 WES是疾病诊断的利器
2009 年8 月,Ng等[19]通过WES从4 例Freeman-Sheldon综合征(FSS)患者的DNA 中准确找出了致病基因MYH3,首次证明了WES在确定罕见疾病致病基因上的应用价值。仅1个月后,Choi等[20]运用WES在先天性氯丢失腹泻患者中发现了新的致病基因突变位点SLC26A3,其研究更重要的意义在于纠正了先前临床的错误诊断,凸显出WES在发现新的致病基因和临床诊断上的准确性。WES正式进入临床则是在2011年,Worthey等[21]对1例难治性炎症性肠病的15月龄男性患儿行WES,在X连锁凋亡抑制基因中鉴定出一个错义突变,并提示脐带血移植治疗策略的可能性。这项研究开创了WES临床诊断的先河,也为个体化医学的发展奠定了基础。
近年来,越来越多的研究者将目光投向WES的临床实践。尤其在罕见性遗传病的研究中显露头角,罕见病多系单基因病。根据OMIM数据库(更新至2014年12月31日),目前世界上报道的已知和怀疑的孟德尔遗传病22 724种,其中确认为孟德尔遗传病且致病分子基础已知的有4 310种,确认为孟德尔遗传病但分子基础未知的1 678种,不确定是否为孟德尔遗传病1 849种。单基因病的早期诊断、预防和治疗,依赖于致病基因的发现、突变谱的鉴定及其功能研究。传统的单基因病致病基因定位方法有候选基因法、定位克隆、全基因组扫描连锁分析、物理图谱、关联分析和全基因组测序(WGS)等技术。然而,这些技术仍然存在难以克服的“瓶颈”,例如疾病遗传异质性和遗传模式的限制,患病亲属人数有限或家系资料不完整,患者为散发性(从头突变)等。因此,至今依然不能完全确定疾病的易感基因。
近年来,越来越多的临床大样本研究表明,WES开始在单基因疾病诊断中显现其独特的价值。Yang等[22]对3 386例患者行WES,830例可明确诊断,诊断率约为25%,进一步验证了该研究团队1年前发表的250例初步报告[23]的诊断率。受试对象大多是儿童,临床表现以神经系统障碍为主,如发育迟缓和智力障碍。其中92例(4.6%)患者可采取有效临床干预措施。Yang等[22]认为,由于WES对外显子区覆盖不完全和对变异的解释不足等局限性,25%的分子诊断率可能是低归因的结果。Lee等[24]对814例患者行3人家系WES(双亲与患儿)和先证者测序,其中520例(64%)是儿童,其诊断率为26%(213/814),而在3人家系测序组诊断率高达31%(127/410),明显高于先证者测序组22%(74/338)的分子诊断率。3人家系WES诊断率高主要是由于对从头突变和杂合子突变的检测能力提高所致。
文献[22,24]为WES临床分子诊断提供了有力证据。但其结果与以12名健康成年人为受试对象探究WGS应用价值的研究不同[25]。原因可能是:①WGS不仅能检测蛋白编码区的基因突变,而且还能检测到非编码区的突变。②文献[22,24,25]验前概率不同:在人群中一个健康个体罹患一种罕见遗传疾病的可能性非常低,而临床试验研究本底人群是患者,每例患者所表现的临床症状很可能就符合一种罕见遗传疾病[26]。总体而言,已报道的WES有害突变检出率在25%[27]~50%[28]。Atwal等[29]向北美47个临床遗传学检测中心发起含8个问题条目的电子问卷调查,得到35份有效WES结果报告,归纳出实际只有22.8%的WES诊断成功率;如果排除可用其他替代方法明确诊断的结果,诊断成功率仅为17.1%;其中,1例还错误地将良性突变诊断为致病性突变。因此,WES实际诊断成功率并不像某些临床实验室和医学文献中报道的那样高,受试人群的病种分布对其有很大影响。Atwal等[29]估计,目前WES有害突变诊断率为15%~25%。
2.1 神经发育异常表型者诊断阳性率高 WES对于神经系统异常表型的诊断率普遍高于非神经系统表型,显示出其在智力障碍和发育迟缓等儿童神经系统疾病诊断中的不俗表现。以往研究显示[30, 31],31%(16/51)的非综合征性智力障碍散发病例和13%(13/100)的严重智力障碍患者可通过新一代测序技术获得特异性分子诊断。Yang等[23]得到25%的WES分子诊断率可能是建立在本底人群临床表型分类不同的基础上,因为250例受试对象中有200例是以智力障碍为临床特征之一。WES对于具有性神经表型的患者(如智力障碍和发育迟缓),分子诊断率高达33%;对于具有特殊神经表型的患者(如运动障碍),WES分子诊断率也达31%。此外,Yang等[22]在更大患者人群中也发现,有特殊的神经系统异常表现组的诊断率高达36.1%,神经系统异常组的诊断率是27.2%,神经系统及其他系统异常组诊断率是24.6%,而非神经系统异常组的诊断率仅为20.1%。在Lee等[24]的表型亚组分析中,发育迟缓的儿童3人家系WES诊断率甚至高达41%,相对于先证者测序9%的诊断率有显著提高。但在心肌病、肿瘤倾向和性发育障碍的表型人群中,3人家系WES诊断率并没有升高。原因可能是医生更愿意将三人WES诊断留给具有复杂综合征的患者,而给非综合征患者选择先证者测序。
WES对于急性和非急性神经发育障碍均有较高敏感度,并且可以缩短患儿诊断时间,指导神经系统疾病的治疗。Soden等[32]对100个家庭的119例患有神经发育障碍[智力障碍、全面发育迟缓和孤独症谱系障碍(ASD)]的儿童及其父母进行WES或WGS,非急性发作表型神经发育障碍患儿WES诊断率40%(34/85),急性发作表型神经发育障碍患儿WGS诊断率 73%(11/15)。Sarah等[32]推测采用WES可使诊断提前77个月,而采用WGS则能再提前10个月。测序分析完成后,近一半家庭的临床诊疗决策发生变化,而有些患者甚至需要改变治疗方式。总体花费也低于以往的个体单个基因测序。可以预见,以基因组为基础的诊断将直接影响患有神经发育障碍儿童的医疗服务模式。

值得注意的是,鉴于目前对通过显性抑制或激活机制起作用的致病突变的检测能力有限,文献[33]显示在无关的3人家系测序组之间观察到4个新基因(COL4A3BP,PPP2R1A,PPP2R5D和PCGF2)具有相同的从头突变。如下2种假说可以解释这一现象:①文献[33]只是在成千上万的不同功能获得性突变中偶然得到了结果,仅触及事实的表面; ②由于这些特殊变异赋予了种系积极的选择优势,因此存在富集现象[37]。评估这些假设将需要更多数据的分析。
由此可见,神经发育障碍表型的患者尤其适合进行WES临床分子诊断,WES是发现及证实许多神经发育疾病致病基因的一种高效方法。WES或WGS将有助于准确描述新发疾病,显示已知疾病不典型或未完全表现出来的表型,并帮助识别共患病。
2.2 能够发现单基因病的复杂表现 文献[23]发现,有4例患者存在一种突变基因对应≥2种疾病状态的现象。纯合子突变引起常染色体隐性遗传的分子机制也多有涉及:59例突变基因分别遗传自父系和母系,5例源自单亲二体,4例则是由杂合子点突变和大范围拷贝数变异(CNVs)的缺失引起。值得注意的是,常染色体隐性遗传模式在获得诊断的患者中所占比例较大(34.3%),这与文献[30]报道不符,100例智力障碍患者WES分子诊断16例,仅有1例是常染色隐性遗传(6.3%)。文献[22]遗传模式分析表明,23例患者(4.6%)是由2个基因缺陷引起的混合表型。文献[33]在17例患儿中鉴定出2个不同基因,均与致病突变有关,从而导致复合临床表型。由此可见,WES能够发现单基因病的复杂表现,随着其在临床上的广泛应用,具有叠加症状和混杂表型的患者的诊断将不再困难。
2.3 对嵌合体的诊断优势明显 如果测序深度高且寻找变异的工具足够敏感,单核苷酸链的单独序列分析能够准确检测嵌合体[39~41]。嵌合体即患者体内仅有一小部分细胞携带突变基因,常是严重早发性疾病的潜在原因,如Cornelia de Lange综合征(CDLS)[42]。CDLS是一种以特殊面容、智力障碍和发育迟缓为特点的多系统疾病。大多数典型CDLS患者在NIPBL有功能丧失性新生杂合突变且嵌合体比例较高。而联合SMC1A、SMC3、HDAC8和RAD21的突变则引起不典型CDLS。Ansari等[43]分别对163、90和19例患者行已知基因编码区的突变、基因重排和NIPBL上深度内含子变异检测,另外对5例行WES,发现致病突变被确定在:NIPBL46(嵌合体3,28.2%);SMC1A5(嵌合体1,3.1%);SMC35(嵌合体1,3.1%);HDAC86(嵌合体0,3.6%);RAD211(嵌合体0,0.6%)。在ANKRD11上鉴定出3个从头突变且与KBG综合征表型重叠。为估计未发现的嵌合体例数,文献[43]使用递归分区来识别NIPBL阳性亚组的不同特点。依据这些特点对突变阴性组过滤,至少有18%被划为“NIPBL样”变异。
Fitzgerald等[33]对1 009例患儿和1 013例对照做外显子组为重点的阵列比较基因组杂交(外显子组-aCGH),1 006例行全基因组基因分型,识别缺失、重复、单亲二体和嵌合体。从WES和外显子组-aCGH的数据发现,每例患儿平均有19 811个编码或拼接的单核苷酸变异(SNVs)、491个编码或indels和148个CNVs。从基因分型阵列的数据[44]分析,文献[33]确定了5例患儿是嵌合的大型染色体重排。文献[22]2 000例患者的遗传模式分析表明,280例是常染色体显性遗传(53.1%),在显性突变中,有208个(87%)是从头突变,其中有5个已证明是嵌合体。由此可见,WES可以检测到低水平的嵌合体,并能更好地估计突变细胞的比例[45~48]。
Petersen等[49]为了明确体细胞突变是否在Crohn病(CD)的发病中起重要作用,对两组同卵双胞胎的肠和血液样本进行WES和WGS,目的是在患病双胞胎中鉴定出候选的新CD易感基因位点。研究表明,体细胞嵌合体似乎并没有在CD同卵双胞胎患者的不一致性中发挥重要作用。但这是首次对CD双胞胎行WGS,因此为今后的研究提供了一个有价值的参考数据集和嵌合体检测的框架。
2.4 可以发现新的致病基因 WES是一个及时更新疾病基因的合适平台。3 386例大样本研究中近30%的诊断基于过去3年内所确定的致病基因。在504例样本中发现了708个候选致病突变,58%的变异未报道过,且除WES外,尚无其他可用的基因检测技术来发现这些未知基因突变[22]。值得注意的是,大部分未诊断病例很可能是由于新致病基因未被发现所致,对疾病相关文献和数据库的定期监测将有助于提高诊断率[50]。de Ligt等[30]研究发现了24个有关智力障碍的从头突变候选基因,其中3个基因的致病作用已被证实; 每1个病例中,携带有同一基因突变的患者间观察到显著的表型重叠现象,所发现的DYNC1H1、GATAD2B和CTNNB1是引起智力障碍的新基因,WES可以作为诊断原因不明的严重智力障碍患者的有效方法; 针对他们所研究的病例,WES诊断率达到16%。随着方法技术的改进以及更多智力障碍相关基因的鉴定,WES诊断率可能还会提高。此外,针对覆盖率低的外显子区,新的靶向捕获试剂正在研发中,以弥补目前对已知疾病基因测序的不足。
WES对新致病基因的发现,为罕见复杂疾病和难诊断疾病的临床处理提供了解决之道。由于多变的外显率、非特异性临床表型或病理学特征,阻碍了Ⅳ型胶原相关肾病的诊断。Lin等[58]对3个伴有原因不明的遗传性肾病家庭行WES,分别鉴定出2个新的和已知的COL4A3/COL4A4/COL4A5致病突变[59~61]。WES为非典型肾病的诊断困境提供了解决之道,同时也使恰当的疾病咨询和治疗建议成为可能。FOXP3突变被认为是IPEX样综合征(immune,dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome)的主要病因,但大型队列研究表明,有>50%的患者未检测到FOXP3突变[62],这为临床提高IPEX样综合征的诊断准确性带来了难题。Charbonnier等[63]试图确定特发性IPEX样综合征患者除FOXP3突变以外的遗传异常,对3个家系进行了WES,并行有针对性的靶基因测序和表型功能分析,结果显示:LRBA基因缺陷是IPEX样综合征和Treg细胞缺乏症的新的致病原因[64~66]。此外,在Yang等[23]的研究中,4例经WES诊断为Noonan综合征,其中3例临床表现不典型;1例表现典型,但基因panel序列分析未发现致病基因,经WES后发现基因CBL上存在有害突变,而此基因未被囊括入panel的新基因。WES不仅可鉴别出新的有害突变,扩充致病基因备选库,而且还能诊断出更多临床表现不典型的常见遗传病,完善疾病表型谱。
基于WES技术的研究设计为鉴定遗传性缺失(missing heritability)提供了新的研究策略[67~69], WES的数据分析可能是一种具有高成本效益的方式,在探索罕见复杂疾病的功能变异和病因学研究中发挥重要作用[70]。
值得注意的是,WES发现的新致病突变有助于进一步划分疾病分子亚型,指导临床靶向治疗。前列腺癌中常见的基因变异包括NKX3.1和PTEN的缺失,雄激素受体基因(AR)的重复,ETS家族转录因子基因和雄激素敏感的启动子序列融合等。以往观点认为频发体细胞碱基对替换较小影响前列腺的肿瘤发生过程[71, 72],但这种观点尚未经大型队列研究证实。Barbieri等[73]对112对前列腺肿瘤/正常组织进行WES,在多个基因中鉴定出新的频发突变,包括MED12和FOXA1。SPOP是突变最频繁的基因[74, 75],有6%~15%的肿瘤组织基因突变涉及SPOP底物结合部位的保守残端[76]。SPOP突变的前列腺癌缺乏ETS重排,并表现出一种独特的基因组改变模式。WES测序结果的SPOP突变可能定义一种新的前列腺癌分子亚型。今后,这一不断扩充的遗传构架会使肿瘤的致癌机制更加明确,有助于疾病建模和患者分层治疗。对前列腺癌的基因组、表观基因组和转录组的系统性综合分析,能够进一步阐明疾病生物学背后的遗传变化,并针对异质性恶性肿瘤的遗传倾向予靶向治疗。
由此可见,WES对新致病基因的发现可能提供病因学研究的新思路。许多研究为不同人类疾病之间的相关性提供了证据,包括孟德尔遗传病和复杂疾病[77~79]、复杂疾病之间的相关性[80]。因此,是否可以提出这样的假设:引起类似临床表型的基因,是能够解释疾病易感性的、有参考价值的候选基因。此外,对涉及这些已知基因的特定细胞通路或网络进行综合分析[81, 82],判断其在患者体内中是否发生结构和功能突变,可以为选择候选基因中的优先考虑基因提供线索。这种方法已在ASD[83]的研究中得到证实,相信在基于WES的研究中也会发挥作用。另外,组合多种预测工具并产生一致性评估得分的方法,已证实有更突出的结果[84, 85]。然而,这些不同的工具或评估得分常常来源或应用于评价特定的数据集,建立一个全面评估敏感度和特异度的无偏倚“金标准”数据集是必要的[57]。
如果WES研究设计能以适当的方式被应用,将会发现更多复杂疾病的新易感基因罕见突变。高质量和高可信度的大型参考数据集将会提高WES研究设计的可行性。包括外显子组芯片在内的其他技术的发展,也在努力为罕见突变的功能注释提供更好的参考数据库。随着进一步的大规模合作,基于病例对照设计的WES研究可能会更加具有可行性。此外,检测新易感基因罕见突变也需要统计学和生物信息学工具的进步。在阐明复杂性状中罕见编码突变的病因学作用后,将有必要使用WGS技术对非编码突变的作用做进一步探讨。合适的统计学和生物信息学分析工具正在积极研发中[86, 87],这对于复杂疾病遗传基础的认识至关重要。而其他探索突变基因功能性和改进功能分析策略的研究,也会使对易感性突变的生物学意义有更深刻的了解[88]。其他数据类型,包括表观遗传变异、mRNA表达水平和体细胞遗传变异等,对考虑优先候选基因是有帮助的,可以为种系遗传变异的疾病转化发生过程提供进一步的深层次理解,为易感基因相关生物学意义提供合理性解释[57]。
3 WES在儿科复杂疾病中的研究应用
复杂疾病是在众多因素共同作用下发生的,如多个基因、一个基因的多个突变、环境作用及未知的随机因素。自2005 年以来,对于复杂疾病的研究,主要采用基于基因芯片的GWAS,通过比较病例与对照间SNP 的频率差异来寻找风险变异因素。然而,目前的GWAS 只涉及复杂疾病和性状易感基因的很少一部分,解释的遗传度极大地低于预期值,并且极少数SNPs被明确具有与疾病机制相关联的功能性作用。基于ASD或精神分裂症的测序研究,证明了对人基因组功能区域进行深度重测序是寻找复杂疾病的潜在罕见致病突变的关键方法[89]。现从儿科疾病的角度出发,主要介绍WES在ASD和先天性心脏病(CHD)中的临床研究现况。
3.1 ASD ASD是一种高度遗传的神经发育紊乱,临床表现以受损的社会互动和交流缺陷,受限制的重复行为为特征[90]。对有ASD患者的家族进行研究发现,儿童ASD同胞患病率为2%~8%,是一般人群发病率的50~200倍。双生子研究则显示,同卵双生的同病一致率为60%,异卵双生的同病一致率为0。这些证据证明遗传因素是儿童ASD发病的重要原因。目前的研究对于候选基因的数目和界定仍不清楚,对存在1例以上ASD患者的家族进行的全基因组分析,估计该病至少是10个以上致病基因相互作用的结果。
随着基因组学研究和测序技术的发展,基因组学已经成为ASD近几年主流的研究方向,每年均有新的ASD相关候选基因被报道。但由于ASD相关突变非常多样,很难建立清晰的遗传模式。大规模平行DNA测序发现了许多与ASD相关的罕见SNVs和微小indels变异。已证实WES对于检测ASD中的从头变异(DNVs)具有独特价值[85,91~93]。WES相关研究已经阐明了罕见遗传性变异(IVs)的作用,如罕见基因缺失(遗传性功能丧失的纯合子,男性复合杂合子或X染色体变异)[94]、隐性纯合子[95]、双等位基因变异[96]和多家系变异共遗传[97]。Chahrour等[95]对163个中东地区的ASD家庭进行WES,发现在基因AMT、PEX7、SYNE1、VPS13B、PAH和POMGNT1上都存在双等位基因突变,此后他们又分析了612个美国ASD家庭的WES数据,得到了相同的发现[96]。这些基因大多与一些代谢或遗传学综合征有关,相关隐性突变影响比较轻微,使蛋白部分丧失功能。Lim等[94]对933例ASD患者的基因组和869例对照进行了WES,以检测使基因功能完全丧失的隐性突变。文献[94,95]研究相互补充,共同说明了隐性突变对ASD的重要性。最近,WGS也已鉴定出与非编码DNA相关的罕见遗传变异和影响基因剪接的变异[98,99]。由此可见,成百上千的罕见遗传变异可以影响与ASD相关的多种生物学功能[100],而以WES为代表的新一代测序技术或许可以成为解开ASD遗传迷雾的突破口。
WES不仅在识别ASD相关致病突变方面作用突显,而且以此为基础推动了ASD发生机制的研究。Willsey等[101]指出了9个高可信度ASD风险基因的功能作用,然后发现所有基因都在胎儿发育的特定时间集中在大脑一个特定场所的一个单细胞类型。De Rubies等[102]和Iossifov等[103]共同合作的2项研究通过WES发现了100多个ASD相关基因突变,大多数都是从头突变,其中60个基因突变有超过90%的概率引发ASD风险。上述研究表明,这些基因中的罕见突变可影响中枢神经网络。An等[104]利用“AXAS”基因网络模型分析从澳大利亚ASD队列中得到的WES数据,该模型描述了在ASD、X-连锁智力障碍、注意缺陷多动障碍和精神分裂症中所占比例过高的异质性DNA变异的功能模式。一种优化的DNA变异筛选流程被用来确定功能缺失的DNA变异。该研究还发现,来自于父母表型更广泛的可遗传变异和从头变异与ASD有显著联系。分析表明,假定罕见致病变异集中位于关键神经生物学过程中的集群和比例大,并且在涉及神经元发育、信号转导和突触发育等功能环节占比过高。这些研究结果同时证实,增加ASD风险的不是突变基因的规模,而是突变基因的位置。这些基因突变或许会导致ASD的风险增加5~20倍。Chang等[105]对数百名患者和近1 000个基因进行大型分析发现,ASD症状的多样性可以追溯到患者基因突变的差异,包括特定基因突变和突变的严重程度,提示可以根据基因型与表型的关系利用患者的遗传图谱,开发精确的诊断和预后工具,甚至引导个性化治疗。
3.2 CHD 是人类最常见的出生缺陷,在所有活产儿中发病率约为1%[106, 107]。大多数CHD是遗传因素与环境相互作用的结果[108]。约30%的心脏畸形是综合征性的疾病,如唐氏综合征、22q11.2缺失综合征和Holt-Oram综合征[109, 110];然而大多数CHD并不遵循孟德尔遗传[111]。
过去10年常采用经典连锁分析或候选基因法探究CHD等家族性疾病的遗传背景,而这些研究数据大多建立在模式生物(如基因敲除小鼠)的基础上[112, 113];染色体畸变(包括CNVs)则通过细胞遗传学分析(如荧光原位杂交FISH等)鉴定[114, 115];比较基因组杂交(aCGH)为筛选亚显微水平染色体不平衡提供了更高的分辨率。利用aCGH的首次在CHD的研究表明,约17%的CHD患者具有潜在致病的罕见染色体畸变[116, 117]。全基因组SNP阵列已被用来确定散发CHD病例中的拷贝数变化[118~120]。为了找到与复杂疾病相关的SNPs,GWAS已在数百到数千人的大型队列研究中展开[121]。相关技术方法也在其他研究中得到证实[122, 123]。总之,上述技术方法为探索CHD的遗传学基础展现了很好的前景。但仍然有很大比例的心脏畸形不能确定遗传病因。
二代测序技术则是目前进一步阐明CHD遗传背景的有效新方法。例如,Dorn等[124]针对CHD提出了一个高通量测序设计和分析路线图,也适用于其他复杂疾病。WES、高分辨率熔炼分析和直接DNA测序的联合应用确定了一个家族异质性CHD可能的致病突变基因[125]。应用WES可以识别出患有先天性心脏病的内脏异位患者SHROOM3上存在错义突变[126]。NGS使大型队列中的成千上万个基因甚至整个基因组实现了同时分析。与微阵列不同,NGS不依赖于DNA杂交预选探针,这有利于在单碱基分辨率上鉴定新变异,这也预示着新疾病基因网络的发现。不过,由于大量数据的产生,要鉴定真正的疾病相关基因是非常复杂的。大规模人口研究表明,可以在任何健康个体中观察到大量潜在的致病变异[127~129]。NGS的运用还发现,一些致病突变在健康个体和个别情况下可以被耐受,尽管发生频率极低。因此,新测序方法应用到类似CHD的复杂疾病分析时仍然具有挑战性。
WES与其他技术的联合应用可弥补其在检测罕见CNVs上的不足,提高对CHD等复杂疾病的应用价值。Glessner等[125]对538例CHD患者和1 301名健康对照采用全基因组SNP微阵列和WES,研究新发CNVs在散发CHD发病中的致病作用;这2种互补的高分辨率技术从51 例CHD患者中鉴定出63个新发CNV,证明CHD患者罕见新发CNV出现频率显著增高:SNP阵列[P=7×10(-5);比值比4.6]或WES[P=6×10(-4);比值比3.5];而除去16%曾被报告为致病性新发CNV位点后,CNV出现频率仍保持较高水平(P=0.02后,比值比为2.7);在15q11.2上的CYFIP1、NIPA1和NIPA2上还检测到频发新发CNV,在DUSP1、JUN、JUP、MED15、MED9、PTPRE、SREBF1、TOP2A和ZEB2上检测到单个新发CNV,均与已知的CHD蛋白NKX2- 5和GATA4存在相互作用;还发现ETS1的11q24.2-25缺失引发Jacobsen综合征,CTBP2是10q端粒缺失的致病基因。
房间隔缺损是CHD最常见的缺陷之一。过去研究表明,转录因子T-box 20突变(TBX20)是先天性房间隔缺损的病因之一。Liu等[126]采用WES联合CHD相关基因过滤检测1家3代的房间隔缺损患者,发现1种新型TBX20突变,c.526G> A(p.D176N)。生物信息学程序(SIFT,Polyphen2和MutationTaster)预测这种突变是有害的。此突变以前并没有包括在SNP数据库(dbSNP)或国家心脏、肺和血液研究所(NHLBI)WES计划(ESP)的数据中。Liu等发现扩大了TBX20突变谱,证明了TBX20在心脏发育中起着重要作用,也为小家系CHD的遗传研究提供了一个性价比较高的新分析策略。Greenway等[130]对1个常染色体显性遗传隔离继发孔型房间隔缺损高发的家系进行WES,此前候选基因测序和连锁分析均无结果。2例患者鉴定出44个罕见共同变异,包括在α-心肌肌动蛋白(ACTC1)中的非同义突变(c.532A> T,p.M178L,NM_005159.4);而1 834名对照、千人基因组和ESP的均无此突变,p.M178L也是唯一1个从家系中分离得到的罕见基因突变。Greenway等[130]的研究为ACTC1突变在房间隔缺损中的致病作用提供了进一步的证据支持,而大规模平行WES也使检测CHD新罕见变异成为可能,不再受到候选基因法的局限。当突变预测算法无效时,家族性疾病的研究可有助于从良性变异中区分罕见的致病突变,重视家族史研究也使对CHD的遗传见解更加深入。
研究证明,SCN5A突变是遗传性心律失常的潜在病因,但其与复杂重叠表型,特别是与CHD的联系则很少报道。Tan等[131]联合运用WES和生物信息学的策略,在1个多代家系中鉴定出常染色体显性心脏传导疾病(CCD)的致病基因,发现SCN5A上存在一种新的无义突变(Y1495X),推测该突变产生截短的SCN5A蛋白从而导致钠电流损失,明确了SCN5A相关性心律失常的机制。由此可见,WES是一种对罕见临床表型做遗传分析的非常有效的方法,为临床表现阴性的家属提供了准确的基因检测信息。
利用高通量测序的大型CHD队列研究有望更好地了解疾病潜在的复杂遗传学基础。儿童心脏基因组协会建立的CHD遗传网络,纳入3 700例各种类型的CHD,旨在探讨CHD患者遗传因素、临床特点和结局之间的关系。正在进行的研究包括对心脏组织样品分别运用WGS和RNA测序以实现CNV的识别,候选基因的重排及体细胞突变和等位基因错误表达的检索[131]。Zaidi等[132]对362例患者及其父母WES从头突变在CHD患者组蛋白修饰基因中呈明显富集现象,说明表观遗传调控在心脏发育中具有重要作用。威康信托基金会桑格研究所发起的解密发育障碍(DDD)研究(http://www.ddduk.org/),从12 000例未确诊的发育障碍儿童及其父母处收集临床数据和DNA样本,其中也包括CHD患者,使用高分辨率aCGH、SNP基因分型和WES来辨别不同疾病潜在的遗传原因。UK10K项目(http://www.uk10k.org/)对125例CHD患者在其罕见疾病样本组内进行WES。后2项研究仍在进行中,尚未公布CHD的相关研究结果。
WES能更好地理解各种综合征和非综合征性CHD的遗传基础[133],但是这些研究能阐明机制的病例比例仍然很小。例如,罕见CNVs能解释5%~20%筛选过的CHD,WES仅能解读10%散发重症CHD病例的从头突变[132,134]。
Blue等[135]研究利用16个有明确CHD病史的家系所组成的队列筛选出57个候选基因的靶向panel,将测序结果分别与15个健康对照、1000WGS计划和全外显子组测序计划中的数据进行比较,31%(5/16)的家系鉴定出致病变异,又在这些致病突变的基础上根据3个家系确定了一个可能的综合征病因;此外,还在25%的家系中发现了意义不明的变异。有人认为,此研究获得基因诊断的疾病,可能仅需临床检查就能明确,例如TFABP2突变(Char综合征),TBX5突变(Holt-Oram综合征)和ELN突变(主动脉瓣上狭窄)。然而现实中,许多患者的临床表型不明显,呈现出外显率下降、多变表型增多的趋势,阻碍了快速便捷的表型识别[136]。而Blue等[135]的研究证明了使用分子诊断能更容易地解决这一临床现实,当然还要考虑到NGS产生了前所未有的海量难题,而表型分型仍然是一个更费时的过程。
从其他对复杂疾病的研究中可以推断,仅靠WES将不能完全确定单一表型特征的CHD的遗传基础,至少与目前WES普遍能检测出致病变异的分析范例不符。例如,Martin等[137]应用WES到二叶主动脉瓣(BAV)及其他心血管畸形高发的17人家系中,评估了3种常用于WES和家系连锁分析的变异选择策略,仍未能确定BAV的遗传变异基础,说明假定致病变异改变编码意义的WGS,可能不足以揭示BAV和其他复杂性状。非编码变异和寡基因遗传可能是这些现象的原因,目前面临的主要挑战将是如何鉴别良性变异和致病突变,并研制出真正个性化的基因检测工具。
从技术角度来看,WES已经取代靶向基因panel,为编码变异提供了一个无偏倚、全基因组范围筛查的平台。然而,其明显优势依赖于耗费巨大的数据分析。另外,WES在覆盖深度方面逊于靶向基因panel,因为NGS需要多个测序读数重叠以提高可信度。因此,WES目前只建议作为当靶向基因panel等未能取得成果时的后续筛查。随着这些技术的日益成熟,类似Blue等[122]研究的意义在于,在不同临床场景下,为不同类型CHD的基因诊断奠定基础。更多的研究者开始转变立场,提出应加强儿科心脏病专家的职业培训和继续医学教育,以适应新技术要求的知识基础和操作能力。核心课程也必须迅速改变,以配合在基因组学领域的进展步伐。
4 WES在儿童肿瘤中的研究应用
4.1 揭示肿瘤发生机制 越来越多的研究者已认识到WES在儿童实体肿瘤发生机制研究中的潜在价值。神经母细胞瘤是儿童最常见的实体肿瘤。为揭示神经母细胞瘤的遗传基础,Sausen等[138]采用大规模并行测序的设计,分别进行了WGS(6例)、WES(16例)、全基因组重排分析(32例)和特定基因位点的靶向分析(40例),鉴定出染色质重塑基因ARID1A和ARID1B突变(8/71,11%),且这些变异均与早期治疗失败和存活率降低有关。根据肿瘤特异性的结构变异,Sausen等[138]设计了鉴定血清中重排DNA片段的新方法,为微小残留病灶的检测和监控提供了个性化的生物学标志物。结果强调了染色质重塑失调在儿童肿瘤发生过程中的重要影响,并为神经母细胞瘤的治疗方案提供了新思路。
肾母细胞瘤是最常见的儿童肾癌。Rakheja等[139]对44例肾母细胞瘤患儿进行WES,发现体细胞microRNA(miRNA)加工酶DROSHA和DICER1存在错义突变,并在MYCN、SMARCA4和ARID1A三个基因中发现新突变。DICER1 RNase IIIB突变优先干扰pre-miRNA发夹结构5′-端的miRNA的加工过程,同时DROSHARNase IIIB突变从整体上通过显性失活机制抑制miRNA的生物合成。而DROSHA和DICER1突变又共同干扰抑癌基因(TSG)miRNA的表达,包括let-7家族、MYCN、LIN28等重要调节因子和其他肾母细胞瘤的原癌基因。由此可见,体细胞miRNA生物合成元件的突变促使miRNA表达重编程,导致肿瘤发生。WES加快了这些新遗传突变的发现速度,为肾母细胞瘤新亚型的划分提供了病因学方面的遗传依据,同时也为机制研究和临床治疗提供了新视角。
胶质瘤是儿童最常见的脑肿瘤。为了确定高级别胶质瘤(HGGs)其他的致病突变,Fontebasso等[140]对543例HGGs患者行WES,在15%的儿童HGGs中,H3K36 三甲基转移酶(trimethyltransferase)SETD2存在突变,且该突变在全基因组范围均显著(FDR=0.029)。其他的队列研究证实,在低级别胶质瘤中未检测到SETD2突变。SETD2是人类唯一的H3K36 三甲基转移酶,而SETD2突变肿瘤显示出H3K36me3水平显著降低(P<0.001),表明该突变是功能损害性的。此外,丧失功能的SETD2突变多发生在年龄较大的儿童和青年人,且特定出现在大脑皮质的HGGs中,与H3.3 G34R/V和IDH突变的情况类似。Fontebasso等[140]研究提示,SETD2突变破坏了组蛋白H3K36位的编码从而导致胶质瘤的发生,WES又从表观遗传学的角度为肿瘤发生机制提供了新思路。
4.2 作为分型依据 肿瘤的分型与其恶性程度有关,不同分型的肿瘤其治疗方式又截然不同,预后也相差甚远。
以往基于活检结果将髓母细胞瘤患者分为标准风险和高风险两类。Pugh等[141]根据基因表达谱和罕见CNVs将髓母细胞瘤分为4种分子亚型,每种亚型具有不同的生存率。他们同时对突变基因进行功能分析,结果表明是由于少数常见基因调控机制受到干扰而引起髓母细胞瘤的发生,这些干扰的形式在各个肿瘤中有所不同,从而进一步揭示了肿瘤异质性的原因。由此可见,不仅需要在基因组水平上对儿童肿瘤进行分层分型,更需要明确哪些基因突变会驱动哪种分子亚型,其机制又如何,从而实现基于分子分型的预后及靶向治疗,提高诊断率和疗效。
4.3 提示肿瘤预后 以儿童常见的室管膜瘤为例,目前的标准治疗包括手术和放射,但化疗效果不佳。Mack等[142]对47例后脑室管膜瘤患儿行WGS和WES,发现基因组突变率极低以及无显著频发体细胞SNVs。虽然缺乏频发的SNVs和集中的罕见CNVs,但预后不良的后脑室管膜瘤表现出CpG岛甲基化表型。CpG甲基化介导的转录沉默区仅仅集中在PRC2复合体的靶基因上,而已知PRC2通过H3K27的三甲基化(H3K27me3)抑制分化基因的表达。CpG岛甲基化阳性表型的后脑室管膜瘤对靶向DNA或H3K27甲基化的体外和体内临床药物有敏感性。表观遗传修饰位点也许可以作为临床化疗的首要候选靶点。同时,这项研究也提示,某些恶性肿瘤可能呈现遗传学改变不显著,而表观遗传学变化明显的特点,而肿瘤预后可能正是与表观遗传学的变化息息相关。
4.4 提供治疗靶点 越来越多的研究和临床试验证实,WES对新致病突变的发现可以为基于全基因组分析技术的治疗干预措施提供潜在的新靶点。
尽管目前转移性或复发性儿童横纹肌肉瘤的生存率有所提高,但患者预后情况仍然不容乐观。横纹肌肉瘤有2种基因表型:PAX3或PAX7融合基因型、缺乏融合基因型。Shern等[143]对147例患者进行了WGS、WES和转录组测序发现,横纹肌肉瘤的体细胞突变率总体比较低,尤其是在含有PAX3/7融合基因的肿瘤中。尽管突变率相对较低,但除了先前报道过的NRAS、KRAS、HRAS、FGFR4、PIK3CA和CTNNB1频发突变外,还发现了新的FBXW7和BCOR频发突变。此外,93%的病例检测到受体酪氨酸激酶/RAS/PIK3CA轴突变,提示基因组学定向疗法可能改善横纹肌肉瘤患者的预后。
颅咽管瘤患者的临床后遗症主要来源于肿瘤浸润或治疗干预使视交叉、垂体柄和下丘脑区域损伤,目前仍缺乏有效的诊断和治疗措施。Brastianos等[144]WES研究发现,CTNNB1突变几乎存在于所有的成釉质细胞型颅咽管瘤中(11/12,92%),而BRAF频发突变存在于所有的乳头状颅咽管瘤(3/3,100%)。靶向基因分型结果与WES测序结果相吻合。CTNNB1和BRAF突变的鉴定为颅咽管瘤的分子治疗提供了重要的靶向位点。
婴儿肌纤维瘤病(IM)是儿童软组织最常见的良性纤维瘤。Martignetti等[145]对9个不同家庭的32个全血样本进行WES发现,PDGFRB[146]和NOTCH3是导致IM的2个关键突变基因。现已有药物(如伊马替尼和舒尼替尼)可抑制PDGFRB和NOTCH3的表达。对PDGFRB和NOTCH3通路之间联系的进一步研究可能会发现导致IM的其他突变,同时对理解肿瘤生长消退机制和寻找靶向治疗位点具有重要意义。
儿童HGGs是一种破坏性极强的恶性肿瘤,诊断后2年存活率<20%,由于其发生机制仍不清楚,故缺乏有效的治疗措施。Wu等[147]分析了127例HGGs患儿,包括弥漫性桥脑内胶质瘤(DIPGs)和非脑干HGGs(NBS-HGGs)。通过WGS、WES和转录组测序,发现只有DIPGs(32%)的ACVR1基因检测到频发体细胞突变。此外,在47%的DIPGs和NBS-HGGs中,因结构变异而产生的融合基因也被检测到,40%的婴儿NBS-HGGs病例中神经营养因子受体基因NTRK1、NTRK2和NTRK3频发融合。值得注意的是,在68%、73%和59%的儿童HGGs中(包括DIPGs和NBS-HGGs),分别发现受体酪氨酸激酶-RAS-PI3K信号通路、组蛋白修饰或染色质重塑过程、以及细胞周期调控通路上都存在靶向突变。某些特定或共同的细胞信号通路内基因位点的突变与儿童HGG有关,提示基于这些位点的靶向治疗具有研究的价值。
骨肉瘤是儿童最常见的原发性骨肿瘤,但30年间其治疗或生存率均无实质性进展。Perry等[148]对59例患儿进行WES、WGS和RNA测序,只有TP53基因在所有样品中有显著突变频率。鉴于肿瘤异质性、基因组不稳定性及大样本获取难度,该研究利用通路分析、shRNA筛选等方法发现,骨肉瘤细胞系对PI3K/mTOR通路的抑制剂敏感。仅仅找到基因突变位点是远远不够的,还需要了解突变基因引起细胞内错误信号传达的机制,并利用药物测试和建立模式动物研究模型[149],才能够为儿童肿瘤的靶向治疗提供更充分的遗传研究基础。
5 WES的应用局限性
Lee等[24]对WES在实际临床研究中的局限性作了总结。①鉴于3人家系WES诊断的高额费用和在无症状父母中检出阳性的可能性,临床医生可能不会选择此种方法。②非随机化分组使未观察到的混杂因素可能影响诊断率。③WES并不能发现所有具有潜在因果关系的基因突变型,可能出现严重遗漏,如病理性重复扩增(见于脊髓小脑共济失调)和大多数罕见CNVs。④WES也不能取样所有的蛋白编码碱基,平均序列覆盖率有限。⑤3人家系WES诊断对表型数据和变异型两者之间联系的解释分析能力上还有待进一步提高。
自美国医学遗传学会(ACMG)公布偶然发现的相关指南后,如何报告WES中预期外发现的问题便引起人们的极大关注[27, 150~153]。ACMG建议,当基于临床目的对儿童或成人开展WGS或WES时,应当评估与24种遗传疾病相关的56个基因[154]。然而WES往往不能产生高质量的结果而错过特定基因中的变异。为此,研究人员调查了56个ACMG基因中突变的假阴性率[153]。对44个外显子组数据集进行了回顾性分析,这些数据来自4个不同的外显子组捕获试剂盒和2个测序平台。研究人员还研究了WES检测56个ACMG基因中相关突变的能力。总体而言,这4种外显子组方法的覆盖度都不足。在每种外显子组方法中,至少有1个基因错过了40%以上的致病基因变异;最差的有4个基因错过了>90%的变异。现有的测序试剂盒有较高的假阴性率。必须强调的是,当临床外显子组分析没有报告致病的基因变异,可能是因为变异所在的区域没有被分析,而不是患者的DNA中没有致病变异。因此,如果外显子组无法获得预期的表现,则应当进一步使用疾病特异的靶向基因panel。产生足够大量的序列数据,以实现最佳的覆盖度。
综上所述,由于WES是将全基因组外显子区域DNA富集后进行高通量测序的基因组分析方法,其对基因组信息的解读可能出现以下重要遗漏:①在染色体末端重复区域内的外显子不能被检测到;②无法发现线粒体基因中的突变;③忽略染色体结构变异,如易位或倒位;④无法有效诊断三碱基突变疾病,如Huntington's病和脆性X综合征;⑤无法发现其他罕见CNVs;⑥内含子中的致病突变会被WES忽略;⑦无法发现单亲二倍体;⑧无法发现调控序列的DNA变异;⑨无法检测基因之间是否有相互作用;⑩无法解读表观遗传学改变。
6 WES的相关伦理问题
目前在遗传学者中已达成这样一种共识:WGS和WES的广泛应用不仅是大势所趋,而且将大有益处[155]。但是,由于其较以前的基因检测相比定位精确度有所下降,对于在测序过程中可能出现的重要临床发现应如何处理,引发了道德辩论。测序的结果可能被指定为有科学意义,也可能恰恰相反。科学意义的结果是指那些有统计证据证明的基因型(遗传变异)与一个特定表型(例如,疾病的症状或危险因素)之间有相互关系的变异。如果没有足够的证据来支持这种关系,那么这种变异叫做“不确定意义变异”(VUS)。VUS并不一定意味着基因型-表型之间的关系不存在,而是表示当前没有该人群足够的统计学证据来证实这个结果。WGS产生大量的数据,许多观察到的变异都只有不确定的意义,需要进一步的临床研究来确定其意义,这也使反馈研究参与者更多个人信息的行为成为收集更多数据的必要(例如,从其他家庭成员采集更多的生物样本和表型数据)。Hallowell等[156]认为,抛开这些发现是有意为之或是偶然出现,如何处理WES和WGS研究的伦理问题也受到临床或科学研究背景的影响。Hallowell等[156]还呼吁,增加样品采集目的的透明度,更加明确科学研究与临床背景转换的议程,落实患者和研究参与者对于偶发问题及其影响的知情权,并晓之以必要的应对措施。
2014年1月,Illumina公司宣布可在不超过1 d的时间内,以1 000美元的梦幻价格完成对1个人的WGS,测序价格的急剧下降无疑极大提高了新一代测序的可行性。虽然从技术上来说测序一个基因组和获得原始数据的成本急剧下降,WGS的总体成本,包括数据的安全储存、解读和处理,目前还不清楚。文献[155]对WGS的成本计算进行了回顾分析,发现如果考虑到分析数据和结果处理时需要的专家团队,就必须认真考虑“1 000美元的基因组”和“100 000美元的分析”。此外,为让临床医生正确解读WGS结果而必需的一般背景知识培训目前还很缺乏[1]。遗传咨询面临的困境在于,向患者解释技术的本质和结果需要大量的时间,而且实际上如何传达信息以让他们更好地理解也是难题所在。目前大多数已构建的引导生物医学研究和诊断实践的伦理框架并非专为NGS的应用而设计[157, 158],因此这些框架可能需要重新阐释和修正。
6.1 知情同意 知情同意实施过程中常存在以下问题:①需告知的信息量超载或信息内容过于复杂;②患者及家属对信息的理解和记忆能力有限;③医患双方间存在信息理解偏差和虚假希望。这些都有可能使WGS实施时知情同意过程变得更加复杂[157, 159, 160]。以下问题尤其需要关注:知情同意过程中哪些信息需要收录、知情同意应如何实现以及是否可以保留在遗传学研究中习惯使用的观念等[161]。主要关注理由之一是最终分析结果的高度不确定性。首先,这种不确定性一方面会造成期望结果的益处和风险因素的不平衡,例如得到不希望得到的信息、丢失隐私权和虚假期望;另一方面,对突变意义的错误理解也间接影响治疗方案[162]。因此,应客观表述WES和WGS在临床应用上的益处[163]。第二,结果的多样性也引出了如下问题:需要决定哪些结果将反馈给研究对象和患者,以及不同的利益相关者在决定反馈给研究对象的结果时有多大的自主权。第三,结果信息可能与家属相关或有暗示作用,而家属并没有参与知情同意过程。第四,考虑到数据的未来利用度和分享度,必须再三斟酌知情同意的适用范围。最后,必须强调知情同意在研究过程和临床场景中的差别,避免治疗性误解的发生。为了在临床中进行WES和WGS时实施恰当的知情同意,应在测序前花费大量时间做充分辅导咨询[164]。具体注意事项可参考Ayuso等[165]总结的WGS知情同意参考标准。
6.2 数据解读 为决定哪些结果需要反馈,Berg等[166]提出了临床WGS中间储仓式系统的构想,根据分类标准决定反馈计划。例如,具有临床可行性的结果一定要上报,无临床意义或不明的结果不需反馈,临床上有效但可行性欠佳的结果根据研究对象的意愿反馈等。McGuire等总结了目前已有的共识:与临床确实相关且具有直接可行性的结果应该反馈给受试对象[167],而不必公开意义不明的研究结果[168]。而对于预期没有估计到的意外结果,或称“脱靶结果”[154],是否应反馈给受试对象至今还没有能被广泛认可的解决方案[154,169~173]。
6.3 数据知情和处理意见 NGS的应用产生了大量的原始序列数据和遗传变异结果:WGS有望揭示300万至400万的基因变异,WES也有望显示大约2万的突变型[164]。Pinxten等[174]分别从数据存储、分析和分享3个方面讨论数据处理的伦理和实用性问题。①是否有及在何种情况下有义务必须做到记录保存,②未成年人达到法定成年年龄时能否访问曾经的测序数据,③寻求新的诊断和治疗方案的患者是否应重新联系[175],④电子病历还不能很好地储存大量WES和WGS原始数据的问题[164],⑤存储的数据可能会产生实际鉴定和破坏隐私保密的风险等[176]。生物信息学分析也并非完全是自动化处理,许多医疗保健机构都缺乏必要的资源投资昂贵的设备,聘请包括生物信息学等方面的有关专家。仍然需要包含更多医疗相关数据的数据库[177]。由此所带来的经济成本的增加可能已超过技术上的花费[178]。此外,决定不同遗传变异对研究对象个人健康的意义[166]和将不同变异恰当分类[176, 179]是很困难的。而要使所得数据价值最大化就必须分享数据,越是更多地分享在表型相似患者身上发现的变异,就越能实现对变异的正确分类。对研究人员和专家们,应提倡并保证数据分享的合理实现[180];对患者和研究对象,应保证他们对数据分享计划的知情,否则缺少透明度会损害公众对WES和WGS的信任[176]。
7 展望
WES的临床价值在于,可对单独依据临床或实验室难以确诊的不典型疾病、检查项目昂贵疾病和基因突变型未知的疾病做出诊断[181],从而更全面地了解疾病的病因和发展史,指导特异性治疗[182]。WES的应用给儿童疾病诊断带来了全新的思路,进一步扩展了人们对疾病谱的认知度。依据目前WES在儿童单基因遗传病和一些复杂疾病研究中的突出表现,不难推测其在肿瘤学、慢性疾病、产前诊断、新生儿疾病筛查和健康人群普查中也将发挥重要作用,因此明确WES的有效应用领域和范围十分必要。
临床医师应该明智地判断在何种情况下需使用WES诊断,向患者及其家属详细解释WES的结果含义、风险和限制,并避免为追求诊断给患者增加不必要的经济负担。WES推动了对疾病遗传致病机制的深入探究,随着致病基因的不断发现和发病机制的逐渐揭示,临床个体化用药和个性化治疗也将真正成为现实,从而显著改善生命质量[183, 184]。
对儿科学领域而言,WES在产前诊断中的意义尤为重要。WES能识别家族中的致病基因携带者和疾病易感者,预测后代患病风险,为早期诊断和排除诊断提供信息基础[185]。患者家属可利用这些信息作为产前诊断、植入前遗传学诊断和是否继续生育的参考,避免严重缺陷患儿的出生或及早采取有效的针对措施。但是,分子诊断的建立能否对患者及其家庭的医疗情况产生深远影响还存在争论。目前的数据依然很难准确识别WES的整体性能[26],由于对突变体致病性的错误解释,以及医学文献中对基因与相关表型的张冠李戴,仍不可避免假阳性的产生。同时,基因病因的未知性和不可测性也增加了假阴性的可能。关于WES性价比、精确度、收益率和与临床基因诊断的有效整合程度还需通过前瞻性研究进一步分析。
如今,WES已成为WGS的经济替代[14],但其在测序覆盖度、检测变异类型、操作复杂度和有效数据的覆盖均一性等许多方面与WGS相比依然存在明显不足。随着WGS的成本不断下降,WES的吸引力将会消退。可以预测,WGS会大范围地替代WES,WES今后可能仅在科研和临床诊断中还有所运用,直至完全退出测序市场。
随着第二代高通量测序技术的广泛应用,其不足之处也日益凸显。例如,读长较短对后续生物信息学分析带来困难[186],扩增后和扩增前DNA 分子片段的数目偏差对基因表达分析有很大影响[187]等。而日渐兴起的第三代单分子测序技术,因其采用单分子读取技术,数据读取速度更快且不需要PCR 扩增,进一步降低了测序成本,比第二代测序技术有着更广阔的应用空间。单分子测序技术的出现将促进个体医学研究的飞速发展。测序通量的增加可以在短时间内就完成人体染色体将近300 亿个碱基对的测序。而费用的降低则使WGS接近甚至达到1 000美元基因组的目标。这预示着建立个人基因组信息档案将不再遥远,个人的基因组基本信息将作为诊断、治疗和预防的手段。
[1]Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health, 2000, 24(3): 164-168
[2]Dawn TM, Barrett JH. Genetic linkage studies. Lancet, 2005, 366(9490): 1036-1044
[3]Teare MD. Approaches to genetic linkage analysis. Methods Mol Biol, 2011, 713: 55-67
[4]Dueker ND, Pericak-Vance MA. Analysis of genetic linkage data for mendelian traits. Curr Protoc Hum Genet, 2014, 83: 1-4
[5]Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature, 2001, 409(6822): 860-921
[6]Cargill M, Altshuler D, Ireland J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet, 1999, 22(3): 231-238
[7]Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol, 2012, 8(12): e1002822
[8]Marian AJ. Molecular genetic studies of complex phenotypes. Transl Res, 2012, 159(2): 64-79
[9]Lunetta KL. Genetic association studies. Circulation, 2008, 118(1): 96-101
[10]Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature, 2009, 461(7265): 747-753
[11]Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics, 2011, 187(2): 367-383
[12]Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science, 2002, 296(5576): 2225-2229
[13]International HapMap Consortium. A haplotype map of the human genome. Nature, 2005, 437(7063): 1299-1320
[14]Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med, 2014, 371(12): 1170
[15]Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: 2008 update. Genome Med, 2009, 1(1): 13
[16]Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A, 2009, 106(45): 19096-19101
[17]Hedges DJ, Burges D, Powell E, et al. Exome sequencing of a multigenerational human pedigree. PLoS One,2009, 4(12): e8232
[18]Jiang X, Chen R, Cheng S, et al. Computational advances in cancer informatics (a). Cancer Inform, 2014, 13(Suppl 1): 45
[19]Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature, 2009, 461(7261): 272-276
[20]Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A,2009, 106(45): 19096-19101
[21]Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med, 2011, 13(3): 255-262
[22]Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA, 2014, 312(18): 1870
[23]Yang Y, Niu Z, Hardison M, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. New Engl J Med,. 2013, 369(16): 1502
[24]Lee H, Deignan JL, Dorrani N, et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA, 2014, 312(18): 1880
[25]Dewey FE, Grove ME, Pan C, et al. Clinical Interpretation and Implications of Whole-Genome Sequencing. JAMA, 2014, 311(10): 1035
[26]Berg JS. Genome-scale sequencing in clinical care: establishing molecular diagnoses and measuring value. JAMA,2014, 312(18): 1865-1867
[27]Eng CM, Yang Y, Plon SE. Genetic diagnosis through whole-exome sequencing. N Engl J Med,2014, 370(11): 1068
[28]Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet, 2012, 49(6): 353-361
[29]Atwal PS, Brennan M, Cox R, et al. Clinical whole-exome sequencing: are we there yet? Genet Med,2014, 16(9): 717-719
[30]de Ligt J, Gilissen C, Del Rosario M, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med, 2012, 367(20): 1921
[31]Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet, 2012, 380(9854): 1674-1682
[32]Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med, 2014, 6(265): 168-265
[33]Fitzgerald TW, Gerety SS, Jones WD, et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature, 2014
[34]Craddock N, Owen MJ. The Kraepelinian dichotomy - going, going... but still not gone. Br J Psychiatry, 2010, 196(2): 92-95
[35]Jacquemont S, Coe BP, Hersch M, et al. A higher mutational burden in females supports a "female protective model" in neurodevelopmental disorders. Am J Hum Genet, 2014, 94(3): 415-425
[36]Levy D, Ronemus M, Yamrom B, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron, 2011, 70(5): 886-897
[37]Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet, 2012, 90(2): 175-200
[38]Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature, 2013, 501(7466): 217-221
[39]Pagnamenta AT, Lise S, Harrison V, et al. Exome sequencing can detect pathogenic mosaic mutations present at low allele frequencies. J Hum Genet,2012, 57(1): 70-72
[40]Tapper WJ, Foulds N, Cross NC, et al. Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PLoS One, 2014, 9(1): e86940
[41]Lohmann K, Klein C. Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics,2014, 11(4): 699-707
[42]Huisman SA, Redeker EJ, Maas SM, et al. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet,2013, 50(5): 339-344
[43]Ansari M, Poke G, Ferry Q, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet,2014, 51(10): 659-668
[44]King DA, Fitzgerald TW, Miller R, et al. A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res,2014, 24(4): 673-687
[45]Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med, 2011,365(7):611-619
[46]Tapper WJ, Foulds N, Cross NC, et al. Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PLoS One,2014, 9(1): e86940
[47]Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science, 2013, 341(6144): 358-359
[48]Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet, 2014, 95(2): 173-182
[49]Petersen BS, Spehlmann ME, Raedler A, et al. Whole genome and exome sequencing of monozygotic twins discordant for Crohn′s disease. BMC Genomics, 2014, 15: 564
[50]Bainbridge MN, Hu H, Muzny DM, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med, 2013, 5(2): 11
[51]Yu S, Chen L, Ye L, et al. Identification of two missense mutations of ERCC6 in three Chinese sisters with cockayne syndrome by whole exome sequencing. PLoS One, 2014, 9(12): e113914
[52]Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev, 2013, 134(5-6): 161-170
[53]Punetha J, Monges S, Franchi ME, et al. Exome sequencing identifies DYNC1H1 variant associated with vertebral abnormality and spinal muscular atrophy with lower extremity predominance. Pediatr Neurol, 2014,pii: S0887-8994
[54]Willemsen MH, Vissers LE, Willemsen MA, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet, 2012, 49(3): 179-183
[55]Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet, 2013, 45(6): 639-647
[56]Fiorillo C, Moro F, Yi J, et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum Mutat, 2014, 35(3): 298-302
[57]Baasch A, Hüning I, Gilissen C, et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia, 2014, 55(4): e25-e29
[58]Lin F, Bian F, Zou J, et al. Whole exome sequencing reveals novel COL4A3 and COL4A4 mutations and resolves diagnosis in Chinese families with kidney disease. BMC Nephrol,2014: 15, 175
[59]Moriniere V, Dahan K, Hilbert P, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol, 2014, 25(12): 2740-2751
[60]Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant, 2013, 28(12): 2946-2960
[61]Fallerini C, Dosa L, Tita R, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet, 2014, 86(3): 252-257
[62]Barzaghi F, Passerini L, Gambineri E, et al. Demethylation analysis of the FOXP3 locus shows quantitative defects of regulatory T cells in IPEX-like syndrome. J Autoimmun,2012, 38(1): 49-58
[63]Charbonnier L, Janssen E, Chou J, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol, 2015, 135(1): 217-227
[64]Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet, 2012, 90(6): 986-1001
[65]Verbsky JW, Chatila T A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr, 2013, 25(6): 708-714
[66]Burns SO, Zenner HL, Plagnol V, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol, 2012, 130(6): 1428-1432
[67]Kiezun A, Garimella K, Do R, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet, 2012, 44(6): 623-630
[68]Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature, 2009, 461(7265): 747-753
[69]Mccarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet, 2008, 9(5): 356-369
[70]Wu L, Schaid DJ, Sicotte H, et al. Case-only exome sequencing and complex disease susceptibility gene discovery: study design considerations. J Med Genet, 2014, 52(1): 10-16
[71]Kumar A, White TA, Mackenzie AP, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A, 2011, 108(41): 17087-17092
[72]Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell, 2010, 18(1): 11-22
[73]Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature Genet, 2012, 44(6): 685-689
[74]Kan Z, Jaiswal BS, Stinson J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature, 2010, 466(7308): 869-873
[75]Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature, 2011, 470(7333): 214-220
[76]Zhuang M, Calabrese MF, Liu J, et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell, 2009, 36(1): 39-50
[77]Blair DR, Lyttle CS, Mortensen JM, et al. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell, 2013, 155(1): 70-80
[78]Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science, 2014, 343(6170): 506-511
[79]Duggan S, Prichard D, Kirca M, et al. Inherited Syndromes Predisposing to Inflammation and GI Cancer. Recent Results Cancer Res, 2011, 185: 35-50
[80]Wu L, Goldstein AM, Yu K, et al. Variants associated with susceptibility to pancreatic cancer and melanoma do not reciprocally affect risk. Cancer Epidemiol Biomarkers Prev, 2014, 23(6): 1121-1124
[81]Iyer G, Al-Ahmadie H, Schultz N, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol, 2013, 31(25): 3133-3140
[82]Jia P, Zhao Z. VarWalker: personalized mutation network analysis of putative cancer genes from next-generation sequencing data. PLoS Comput Biol, 2014, 10(2): e1003460
[83]O′Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature,2012, 485(7397): 246-250
[84]Li MX, Kwan JS, Bao SY, et al. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet, 2013, 9(1): e1003143
[85]Bendl J, Stourac J, Salanda O, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Bio, 2014, 10(1): e1003440
[86]Torkamani A, Scott-Van ZA, Topol EJ, et al. Annotating individual human genomes. Genomics,2011, 98(4): 233-241
[87]Khurana E, Fu Y, Colonna V, et al. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science, 2013, 342(6154): 1235587
[88]Do R, Kathiresan S, Abecasis GR. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum Mol Genet, 2012, 21(R1): R1-R9
[89]Myers RA, Casals F, Gauthier J, et al. A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet, 2011, 7(2): e1001318
[90]Cristino AS, Williams SM, Hawi Z, et al. Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol Psychiatry,2014, 19(3): 294-301
[91]Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 2012, 485(7397): 242-245
[92]Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 2012, 485(7397): 237-241
[93]Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron, 2012, 74(2): 285-299
[94]Lim E T, Raychaudhuri S, Sanders SJ, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron, 2013, 77(2): 235-242
[95]Chahrour MH, Yu TW, Lim ET, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet, 2012, 8(4): e1002635
[96]Yu TW, Chahrour MH, Coulter ME, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron, 2013, 77(2): 259-273
[97]Toma C, Torrico B, Hervas A, et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol Psychiatry, 2014, 19(7): 784-790
[98]Jiang YH, Yuen RK, Jin X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet, 2013, 93(2): 249-263
[99]Shi L, Zhang X, Golhar R, et al. Whole-genome sequencing in an autism multiplex family. Mol Autism, 2013, 4(1): 8
[100]Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res, 2011, 1380: 42-77
[101]Willsey AJ, Sanders SJ, Li M, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 2013, 155(5): 997-1007
[102]De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature,2014, 515(7526): 209-215
[103]Iossifov I, O′Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature,2014, 515(7526): 216-221
[104]An JY, Cristino AS, Zhao Q, et al. Towards a molecular characterization of autism spectrum disorders: an exome sequencing and systems approach. Transl Psychiatry, 2014, 4: e394
[105]Chang J, Gilman SR, Chiang AH, et al. Genotype to phenotype relationships in autism spectrum disorders. Nat Neurosci, 2015, 18(2): 191-198
[106]Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol, 2002, 39(12): 1890-1900
[107]Reller MD, Strickland MJ, Riehle-Colarusso T, et al. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J Pediatr, 2008, 153(6): 807-813
[108]Nora JJ. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation, 1968, 38(3): 604-617
[109]Antonarakis SE, Lyle R, Dermitzakis ET, et al. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet, 2004, 5(10): 725-738
[110]Momma K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol, 2010, 105(11): 1617-1624
[111]Basson CT, Bachinsky DR, Lin RC, et al. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet, 1997, 15(1): 30-35
[112]Schott JJ, Benson DW, Basson CT, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science,1998, 281(5373): 108-111
[113]Sperling S, Grimm CH, Dunkel I, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat, 2005, 26(6): 575-582
[114]Driscoll DA, Salvin J, Sellinger B, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet, 1993, 30(10): 813-817
[115]Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med, 2003, 9(9): 383-389
[116]Thienpont B, Mertens L, de Ravel T, et al. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J, 2007, 28(22): 2778-2784
[117]Erdogan F, Larsen LA, Zhang L, et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet, 2008, 45(11): 704-709
[118]Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet,2011, 12(5): 363-376
[119]Greenway SC, Pereira AC, Lin JC, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet, 2009, 41(8): 931-935
[120]Soemedi R, Wilson IJ, Bentham J, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet, 2012, 91(3): 489-501
[121]Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med, 2010, 363(2): 166-176
[122]Blue GM, Kirk EP, Sholler GF, et al. Congenital heart disease: current knowledge about causes and inheritance. Med J Aust, 2012, 197(3): 155-159
[123]Fahed AC, Gelb BD, Seidman JG, et al. Genetics of congenital heart disease: the glass half empty. Circ Res, 2013, 112(4): 707-720
[124]Dorn C, Grunert M, Sperling SR. Application of high-throughput sequencing for studying genomic variations in congenital heart disease. Brief Funct Genomics, 2014, 13(1): 51-65
[125]Glessner JT, Bick AG, Ito K, et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res, 2014, 115(10): 884-896
[126]Liu JJ, Fan L L, Chen JL, et al. A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect. J Zhejiang Univ Sci B, 2014, 15(9): 830-837
[127]Tennessen JA, Bigham AW, O′Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science,2012, 337(6090): 64-69
[128]Marth GT, Yu F, Indap AR, et al. The functional spectrum of low-frequency coding variation. Genome Biol,2011, 12(9): R84
[129]Li Y, Vinckenbosch N, Tian G, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet, 2010, 42(11): 969-972
[130]Greenway SC, Mcleod R, Hume S, et al. Exome sequencing identifies a novel variant in ACTC1 associated with familial atrial septal defect. Can J Cardiol, 2014, 30(2): 181-187
[131]Tan ZP, Xie L, Deng Y, et al. Whole-exome sequencing identifies Y1495X of SCN5A to be associated with familial conduction disease and sudden death. Sci Rep, 2014, 4: 5616
[132]Zaidi S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature, 2013, 498(7453): 220-223
[133]Wessels MW, Willems PJ. Genetic factors in non-syndromic congenital heart malformations. Clin Genet, 2010, 78(2): 103-123
[134]Soemedi R, Wilson IJ, Bentham J, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet,2012, 91(3): 489-501
[135]Blue GM, Kirk EP, Giannoulatou E, et al. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol,2014, 64(23): 2498-2506
[136]Andelfinger G. Next-generation sequencing in congenital heart disease: do new brooms sweep clean? J Am Coll Cardiol,2014, 64(23): 2507-2509
[137]Martin LJ, Pilipenko V, Kaufman KM, et al. Whole exome sequencing for familial bicuspid aortic valve identifies putative variants. Circ Cardiovasc Genet,2014, 7(5): 677-683
[138]Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet, 2013, 45(1): 12-17
[139]Rakheja D, Chen KS, Liu Y, et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun, 2014, 2: 4802
[140]Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol, 2013, 125(5): 659-669
[141]Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature, 2012, 488(7409): 106-110
[142]Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. 2014: 506, 445-450
[143]Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov, 2014, 4(2): 216-231
[144]Brastianos PK, Taylor-Weiner A, Manley PE, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nature Genet,2014, 46(2): 161-165
[145]Martignetti JA, Tian L, Li D, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet, 2013, 92(6): 1001-1007
[146]Cheung YH, Gayden T, Campeau PM, et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet,2013, 92(6): 996-1000
[147]Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet, 2014, 46(5): 444-450
[148]Perry JA, Kiezun A, Tonzi P, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A, 2014, 111(51): E5564-E5573
[149]Majczenko K, Davidson AE, Camelo-Piragua S, et al. Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am J Hum Genet, 2012, 91(2): 365-371
[150]Green RC, Lupski JR, Biesecker LG. Reporting genomic sequencing results to ordering clinicians: incidental, but not exceptional. JAMA, 2013, 310(4): 365-366
[151]Ross LF, Rothstein MA, Clayton EW. Mandatory extended searches in all genome sequencing: "incidental findings," patient autonomy, and shared decision making. JAMA,2013, 310(4): 367-368
[152]Klitzman R, Appelbaum PS, Chung W. Return of secondary genomic findings vs patient autonomy: implications for medical care. JAMA,2013, 310(4): 369-370
[153]Park JY, Clark P, Londin E, et al. Clinical exome performance for reporting secondary genetic findings. Clin Chem, 2015,61(1):213-220
[154]Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med, 2013, 15(7): 565-574
[155]Lerner-Ellis JP. The clinical implementation of whole genome sequencing: a conversation with seven scientific experts. J Inherit Metab Dis,2012, 35(4): 689-693
[156]Hallowell N, Hall A, Alberg C, et al. Revealing the results of whole-genome sequencing and whole-exome sequencing in research and clinical investigations: some ethical issues. J Med Ethics,2014,pii: medethics-2013-101996
[157]Caulfield T, Mcguire AL, Cho M, et al. Research ethics recommendations for whole-genome research: consensus statement. PLoS Biol, 2008, 6(3): e73
[158]Kaye J, Boddington P, de Vries J, et al. Ethical implications of the use of whole genome methods in medical research. Eur J Hum Genet, 2010, 18(4): 398-403
[159]Levenseller BL, Soucier DJ, Miller VA, et al. Stakeholders′ opinions on the implementation of pediatric whole exome sequencing: implications for informed consent. J Genet Couns, 2014, 23(4): 552-565
[160]Mcguire AL, Caulfield T, Cho MK. Research ethics and the challenge of whole-genome sequencing. Nat Rev Genet, 2008, 9(2): 152-156
[161]Ormond KE. From genetic counseling to "genomic counseling". Mol Genet Genomic Med, 2013, 1(4): 189-193
[162]Hayden EC. Sequencing set to alter clinical landscape. Nature,2012, 482(7385): 288
[163]Brunham LR, Hayden M R. Medicine. Whole-genome sequencing: the new standard of care?. Science, 2012, 336(6085): 1112-1113
[164]Johansen TK, Dickinson BD, Wilson M. The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med, 2014, 174(2): 275-280
[165]Ayuso C, Millan JM, Mancheno M, et al. Informed consent for whole-genome sequencing studies in the clinical setting. Proposed recommendations on essential content and process. Eur J Hum Genet, 2013, 21(10): 1054-1059
[166]Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med, 2011, 13(6): 499-504
[167]Wolf SM. Return of individual research results and incidental findings: facing the challenges of translational science. Annu Rev Genomics Hum Genet, 2013, 14: 557-577
[168]Mcguire AL, Lupski JR. Personal genome research : what should the participant be told? Trends Genet,2010, 26(5): 199-201
[169]van El CG, Cornel MC, Borry P, et al. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet, 2013, 21(6): 580-584
[170]Miller FA, Giacomini M, Ahern C, et al. When research seems like clinical care: a qualitative study of the communication of individual cancer genetic research results. BMC Med Ethics, 2008, 9: 4
[171]Murphy J, Scott J, Kaufman D, et al. Public expectations for return of results from large-cohort genetic research. Am J Bioeth, 2008, 8(11): 36-43
[172]Shalowitz DI, Miller FG. Communicating the results of clinical research to participants: attitudes, practices, and future directions. PLoS Med, 2008, 5(5): e91
[173]Kaufman D, Murphy J, Scott J, et al. Subjects matter: a survey of public opinions about a large genetic cohort study. Genet Med, 2008, 10(11): 831-839
[174]Pinxten W, Howard HC. Ethical issues raised by whole genome sequencing. Best Practice & Research Clinical Gastroenterology, 2014, 28(2): 269-279
[175]Jacob HJ, Abrams K, Bick DP, et al. Genomics in clinical practice: lessons from the front lines. Sci Transl Med,2013, 5(194): 194-195
[176]Tabor HK, Berkman BE, Hull SC, et al. Genomics really gets personal: how exome and whole genome sequencing challenge the ethical framework of human genetics research. Am J Med Genet A,2011, 155A(12): 2916-2924
[177]Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2013, 42(D1): D980-D985
[178]Mardis ER. The $1,000 genome, the $100,000 analysis?. Genome Med, 2010, 2(11): 84
[179]Grove ME, Wolpert MN, Cho MK, et al. Views of genetics health professionals on the return of genomic results. J Genet Couns, 2014, 23(4): 531-538
[180]Mabile L, Dalgleish R, Thorisson GA, et al. Quantifying the use of bioresources for promoting their sharing in scientific research. Gigascience, 2013, 2(1): 7
[181]Johansen TK, Dickinson BD, Wilson M. The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med, 2014, 174(2): 275-280
[182]Desai AN, Jere A. Next-generation sequencing: ready for the clinics? Clin Genet, 2012, 81(6): 503-510
[183]Fan Z, Greenwood R, Felix AC, et al. GCH1 heterozygous mutation identified by whole-exome sequencing as a treatable condition in a patient presenting with progressive spastic paraplegia. J Neurol, 2014, 261(3): 622-624
[184]Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med, 2011, 13(3): 255-262
[185]Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med, 2014, 16(12): 922-931
[186]Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet,2008, 24(3): 142-149
[187]Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, et al. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol, 2005, 71(12): 8966-8969
(本文编辑:张崇凡)
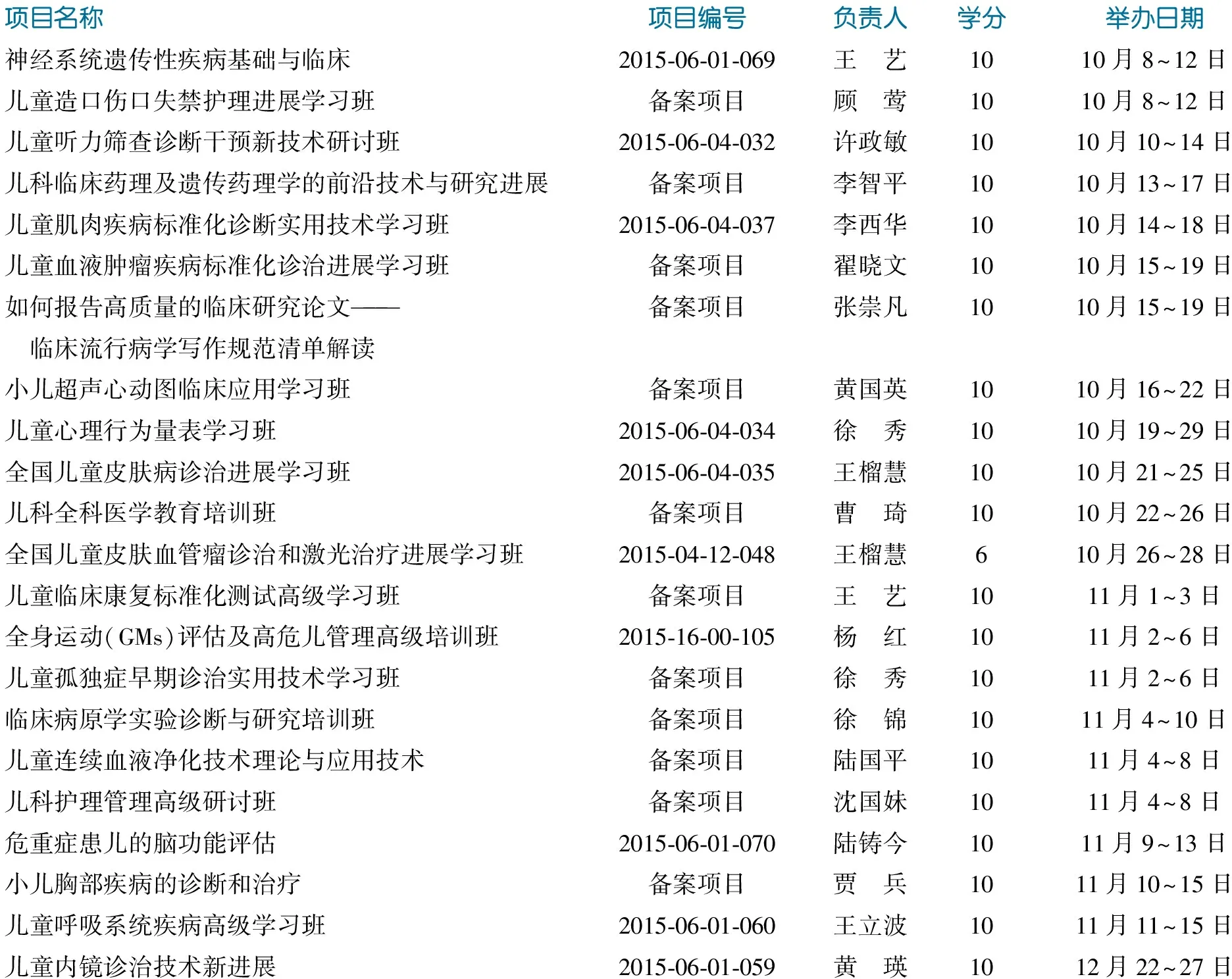
复旦大学附属儿科医院2015年国家级继续医学教育项目(二)
10.3969/j.issn.1673-5501.2015.01.002
上海市卫生局重要疾病攻关项目:2013ZYJB0015;上海市科委/医学领域重点项目子课题:14411950402,14DJ1400103;上海市卫计委项目:沪卫计科教〔2013〕018号
1 复旦大学附属儿科医院 上海,201102;2 上海市出生缺陷防治重点实验室,复旦大学儿童发育与疾病转化医学研究中心,卫生部新生儿疾病重点实验室,复旦大学附属儿科医院上海,201102;3 复旦大学生物医学研究院上海,200032;4 共同第一作者
周文浩,E-mail:zwhchfu@126.com
2014-12-17
2015-01-20)
