高通量测序数据分析和临床诊断流程对新生儿多发畸形候选变异的筛选准确性研究
2015-04-21黎籽秀王慧君周文浩
黎籽秀 刘 博 杨 琳 王慧君 周文浩
·论著·
高通量测序数据分析和临床诊断流程对新生儿多发畸形候选变异的筛选准确性研究
黎籽秀1,5刘 博2,5杨 琳3王慧君4周文浩4
目的 构建应用于遗传性疾病诊断的基因组二代测序数据分析流程,为候选变异筛选提供参考流程。方法 复旦大学附属儿科医院(我院)新生儿出生缺陷生物样本库中的87例多发畸形样本,以美国贝勒外显子组测序分析流程(贝勒流程)为参考标准进行候选变异筛选,同一批样本以我院建立的全外显子组测序分析流程(复旦流程)也行候选变异筛选,以染色体的位置和突变的碱基作为突变位点的标识符对贝勒和复旦流程检出变异逐行比较。结果 87例新生儿多发畸形样本共检出变异7 820 777个,复旦流程经质量控制、公共数据库的变异频率筛选、内部数据库的变异频率筛选、变异类型的筛选和关注与孟德尔遗传病相关的变异数筛选,平均单个病例180~210个变异作为最终候选变异行临床诊断分析;贝勒流程检出候选变异15 242个,复旦流程检出候选变异15 660个,2个流程均检出候选变异15 137个,复旦流程比贝勒流程多检出的候选变异523个,105个候选变异贝勒流程检出复旦流程未检出,复旦流程敏感度、特异度、阳性预测值和阴性预测值均高于96.66%。结论 虽然复旦流程候选变异筛选能真实有效地模拟贝勒流程对WES数据进行筛选,并寻找候选变异的过程;复旦流程所展现的注释和筛选只是粗略的框架,但提供了WES应用于临床时不可或缺的筛选程序。
全外显子组测序; 新生儿; 敏感度; 特异度; 筛选流程; 多发畸形
基因组二代测序技术与产生低通量数据的传统分子检测技术不同,可以同时对大量基因进行检测,一次检测即可获得海量的变异数据。因此,构建一个基于遗传性疾病诊断需要的基因组二代测序数据分析流程,以期从众多变异中筛选出潜在致病突变显得尤为重要。
目前,已有许多权威科研机构开设以全外显子组测序技术(WES)来发现罕见病致病基因的项目,如加拿大罕见疾病基因发现中心项目[1],英国国民保健服务NHS和Sanger研究院合作项目[2,3],美国国立卫生研究院的未确诊疾病计划[4]等。这些项目或计划在过去的2~3年中,基于各自的研究需要,许多实验室都建立了相应的基于遗传性疾病诊断需要的基因组二代测序数据分析流程。虽然各流程的建立均是为了聚焦到候选致病变异,但每个流程对变异的注释方式和筛选方法各有不同。美国贝勒医学院人类与分子遗传系是第一个以方法学的形式展示WES用于临床的诊断流程(简称贝勒流程)的实验室,贝勒流程对2011年10月至2012年6月收集的250例未经选择的患者进行外显子组测序,并成功明确了其中25%的患儿存在的致病突变[5],并在后续基于贝勒流程的研究,扩大至2 000例病例的诊断率为25.2%[6]。基于遗传性疾病诊断需要的基因组二代测序数据分析流程的建立为WES转化到临床起到了重要的作用。
复旦大学附属儿科医院(我院)转化中心在参考学习了贝勒流程的基础上,建立了高通量测序数据分析和临床诊断流程(简称复旦流程)。本研究以我院新生儿出生缺陷生物样本库中多发畸形患儿为样本,以贝勒流程检出候选变异为参考标准,采用同一批病例以复旦流程进行重复检测,检验复旦流程与贝勒流程在候选变异的检出方面的一致性。
1 方法
1.1 检测样本来源 取自我院新生儿出生缺陷生物样本库中的多发畸形样本,多发畸形:≥2项畸形表型。①神经系统畸形:颅骨畸形、脑发育不良、不明原因惊厥和(或)脑电图异常、不明原因肌张力增高或低下、先天性脑积水、不明原因颅内占位和(或)出血;②循环系统畸形:先天性心脏病(单纯动脉导管>5 mm)、心脏不明原因占位;③泌尿系统畸形:肾脏发育异常、输尿管发育异常、尿道下裂;④消化系统发育异常:食道闭锁和(或)气管食管瘘、胆道闭锁、肠道畸形、肛门闭锁、腹壁裂、腹壁疝;⑤呼吸系统发育异常:后鼻孔闭锁、先天性喉软骨软化、肺发育异常;⑥骨骼发育异常:脊柱和(或)四肢骨骼畸形、关节畸形;⑦指/趾畸形:指/趾数量和形态异常;⑧颜面及附属器官异常:眼、耳、鼻、口、舌、腭、颈、毛发异常;⑨生殖系统异常:两性畸形、隐睾。
1.2 贝勒流程 本研究以贝勒流程为参考标准,理由如下:①贝勒流程对WES数据处理并用于临床诊断的结果获得了美国病理学研究院和临床实验室改进修正案(CLIA)的认证[7],是目前公认的WES数据分析的经典流程。②本研究实施贝勒流程行候选变异筛选的工作,是由具有丰富工作和科研经验的贝勒实验室工作人员在我院转化医学中心,以我院新生儿生物样本库中的多发畸形样本在晚近1年时间内完成的。
1.3 复旦流程图1为参考了贝勒流程和相关文献建立的复旦流程,图1中
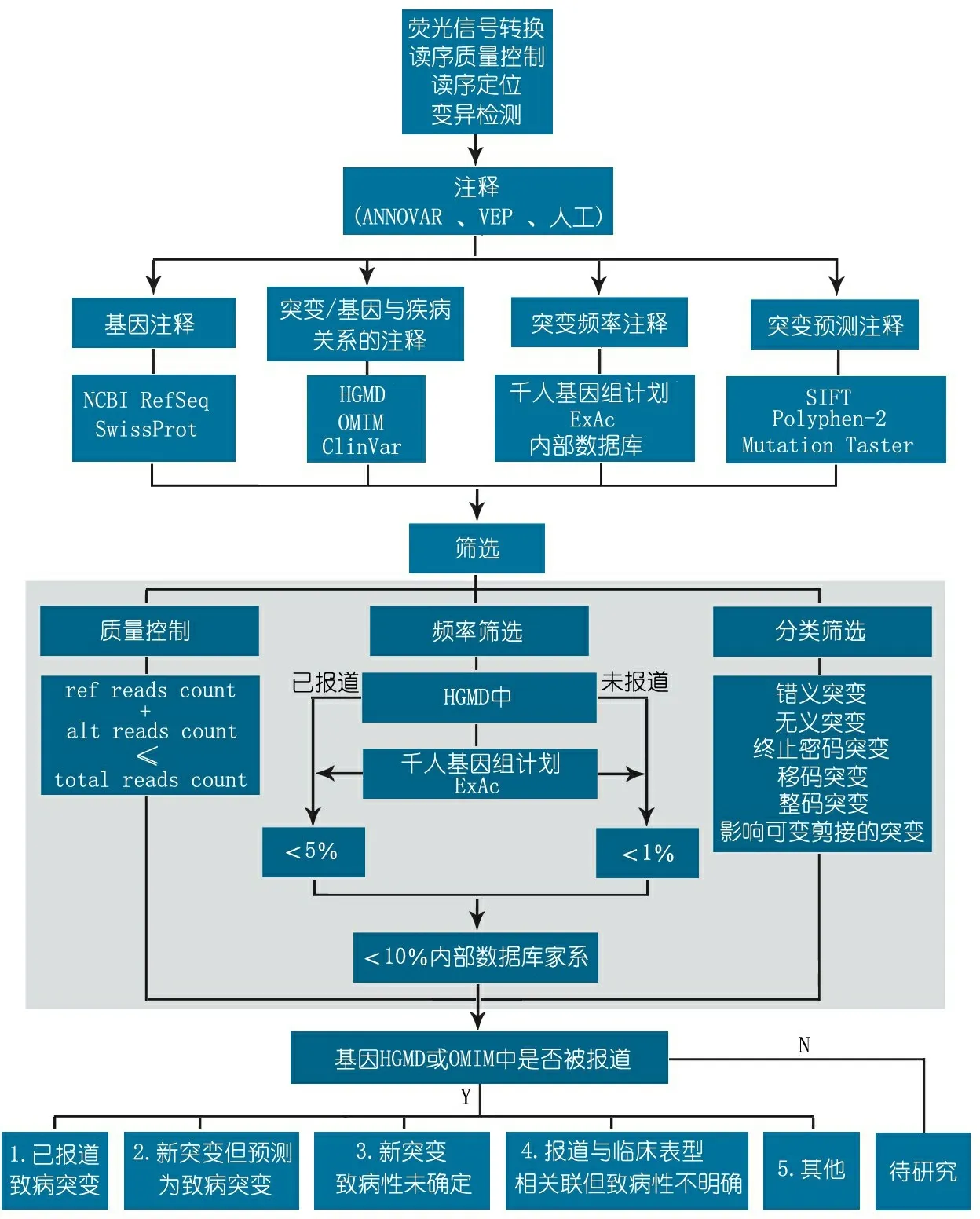
图1 高通量测序数据分析和临床诊断流程
Fig 1 Flow chart of the diagnosis of hereditary disease based on next-generation-sequencing
注释过程通过ANNOVAR(http://www.openbioinformatics.org/annovar/)、VEP(Variant Effect Predictor,http://asia.ensembl.org/info/docs/tools/vep/index.html)软件和人工注释完成;基因注释参考数据库为NCBI RefSeq(NCBI The Reference Sequence: http://www.ncbi.nlm.nih.gov/refseq/)和SwissProt(http://www.uniprot.org/);突变/基因与疾病关系注释参考数据库为HGMD(The Human Gene Mutation Database,http://www.hgmd.cf.ac.uk/ac/index.php)、OMIM(Online Mendelian Inheritance in Man,http://omim.org/)和 ClinVar(http://www.ncbi.nlm.nih.gov/clinvar/);突变频率注释参考数据库为千人基因组计划(1000 Genome Project,http://www.1000genomes.org/)、ExAc(The Exome Aggregation Consortium, http://exac.broadinstitute.org/)和内部数据库;突变预测注释参考数据库为SIFT(Sorting Intolerant From Tolerant)、Polyphen-2(Polymorphism Phenotyping v2)和MutationTaster(http://www.mutationtaster.org/)。
复旦流程在贝勒流程基础上主要做出如下调整:①变异注释的公共数据库为ExAC(http://exac.broadinstitute.org/)数据库[8]。②使用我院转化医学中心的内部数据库,其频率筛选的阈值设置为10%。
1.4 数据对比 本文只对2个流程最后检出的候选致病变异进行比较。以染色体位置和突变碱基作为突变位点的标识符,采用Perl程序比对两个流程处理后所得候选致病变异的异同。
1.5 统计学分析 以贝勒流程检出候选变异为参考标准,真阳性:贝勒流程与复旦流程均阳性;假阳性:复旦流程阳性,贝勒流程阴性;真阴性:贝勒流程与复旦流程均阴性;假阴性:贝勒流程阳性,复旦流程阴性。计算敏感度、特异度、阳性预测值、阴性预测值及其95%CI。
2 结果
2.1 一般情况 共纳入87例多发畸形新生儿样本,神经系统畸形32例次(36.8%),呼吸系统畸形28例次(32.2%),循环系统畸形26例次(29.9%),消化系统畸形25例次(28.7%),四肢畸形25例次(28.7%),特殊面容24例次(27.6%),指/趾畸形10例次(11.5%),泌尿生殖系统畸形7例次(8.0%),骨骼及脊柱畸形5例次(5.7%)。
2.2 复旦流程对变异筛选 表1显示,①质量控制, SNP筛除掉0个变异,插入缺失变异(Indel)筛除约50%(481 502/1 011 134);②变异频率筛选:通过公共数据库筛选后约60%(5 020 513/7 820 777)变异,以内部数据库进一步筛选,筛除约1%的变异[(5 020 513-4 961 147)/5 020 513)];③变异类型筛选,569~790个变异被保留;④关注与孟德尔遗传病相关的变异,134~210个变异被保留
表1 87例新生儿多发畸形复旦流程候选变异筛选(个)
Tab 1 The candidate variants of multiple malformation screening in 87 neonates by Fudan process
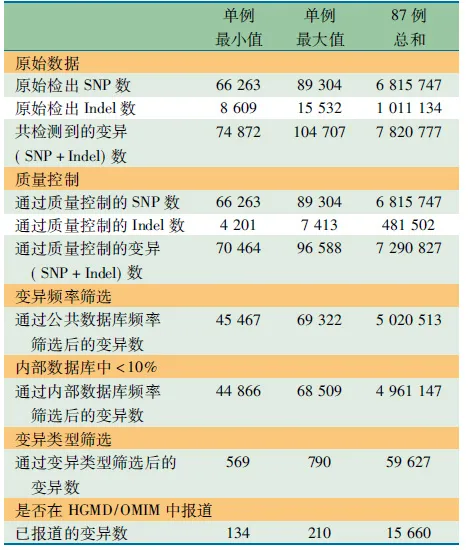
单例最小值单例最大值87例总和原始数据原始检出SNP数66263893046815747原始检出Indel数8609155321011134共检测到的变异(SNP+Indel)数748721047077820777质量控制通过质量控制的SNP数66263893046815747通过质量控制的Indel数42017413481502通过质量控制的变异 (SNP+Indel)数70464965887290827变异频率筛选通过公共数据库频率 筛选后的变异数45467693225020513内部数据库中<10%通过内部数据库频率 筛选后的变异数44866685094961147变异类型筛选通过变异类型筛选后的 变异数56979059627是否在HGMD/OMIM中报道已报道的变异数13421015660
作为最终的候选变异用于临床诊断分析。87例新生儿多发畸形中,平均初始检测出89 894个变异,经由复旦流程筛选,最终检出候选变异平均180个。
2.3 复旦流程与贝勒流程筛选验证 87例多发畸形新生儿样本中,深圳华大基因的数据共检测到变异数目7 820 777个,经贝勒流程检出候选变异15 242个,经复旦流程检出候选变异15 660个,根据染色体的位置和突变的碱基作为突变位点标识符的比对结果,贝勒流程和复旦流程均检出的候选变异15 137个,复旦流程比贝勒多检出的候选变异523个,有105个候选变异贝勒流程检出但复旦流程未检出,表2显示复旦流程敏感度99.31%(95%CI:99.17~99.43),特异度99.99%(95%CI:99.99~99.99),阳性预测值96.66%(95%CI:96.37~96.93),阴性预测值99.99%(95%CI:99.99~99.99)。
表2 复旦流程对新生儿多发畸形候选变异筛选的诊断参数
Tab 2 Diagnostic accuracy assessment of Fudan process for the screening of multiple malformation in neonates

复旦流程真阳性(n)15137假阳性(n)523假阴性(n)105真阴性(n)7805117敏感度/%(95%CI)99.31(99.17~99.43)特异度/%(95%CI)99.99(99.99~99.99)阳性预测值/%(95%CI)96.66(96.37~96.93)阴性预测值/%(95%CI)99.99(99.99~99.99)
3 讨论
本研究以贝勒流程对多发畸形新生儿样本检出候选变异为参考标准,以验证复旦流程检出候选变异准确性。①美国贝勒医学院人类与分子遗传系应用贝勒流程对250例和2 000例孟德尔遗传病行WES数据分析结果先后发表在NEnglMedJ和JAMA,是一个将在人类基因组计划实施过程中积累的知识和技术转换到临床找寻候选致病变异的经典流程[9]。②本研究多发畸形新生儿样本是由贝勒实验室具有丰富的WES工作经验和科研经验的专家依照贝勒流程行候选变异筛选,最终临床诊断率为25%,与贝勒实验室既往研究一致。
建立复旦流程的小组成员由生物信息学背景的分析员和医学遗传学背景的遗传专科医生共同组成。小组成员曾赴美国贝勒医学院人类分子遗传系进行学习和交流,并一直在具有丰富工作、科研经验的贝勒实验室工作人员的指导下,逐步掌握这一领域及贝勒分析流程。
复旦流程在参考学习了贝勒流程基础上做出了2个方面的调整。第一,变异注释使用的公共数据库贝勒流程选用的是ESP4500(http://evs.gs.washington.edu/EVS)数据库,复旦流程选用的是ExAC数据库。ExAC是专门研究外显子组测序数据的联盟机构,该数据库整合了包括ESP4500的数据在内的多个外显子组测序计划,包括了61 486个独立样本的数据,能为样本提供更大规模的对照群体;ExAC是目前收录不包含严重儿童疾病样本的最大数据库,能更好地作为研究儿童孟德尔遗传病的合理对照,较符合我院的实际情况。第二,内部数据库频率筛选的阈值贝勒流程为2%,复旦流程设置为10%。主要体现在:①我院数据库收录的总样本量较小且无关样本量较少,在频率筛选过程中为了保证变异在内部数据中具有一定的构成比而适当做出了调整。②数据库收录疾病种类多样性不够。新生儿样本库在收集样本的时候有偏好性,即对感兴趣的疾病收录样本较多,而对于不感兴趣的疾病则未将其收录,导致数据库中的样本疾病种类较少,存在同一个位点导致多个患者患病的可能。反观贝勒实验室的内部数据库,其收录样本数量大,种类多,同一个位点导致多个患者患病的可能性很小。
通过与贝勒流程比较,复旦流程候选变异筛选敏感度、特异度、阳性预测值和阴性预测值均≥96.66%,能真实有效地模拟贝勒流程对WES数据进行筛选和寻找候选变异的过程,为我院后续的分子遗传学诊断提供保障。
本文对比贝勒流程和复旦流程对新生儿多发畸形候选变异的筛选,只是对这些多发畸形新生儿进行诊断的基础性工作的一部分,不论是贝勒流程还是复旦流程最终明确致病突变还需要对候选变异做进一步评估。复旦流程所展现的注释和筛选只是粗略的框架,但提供了WES应用于临床诊断时不可或缺的筛选程序。
本文有105个候选变异经贝勒流程筛选得到,而复旦流程筛选未检出,分析原因与使用的不同的内部数据库有关。复旦流程用我院转化医学中心内部数据库能够筛除一些贝勒内部数据库不能筛除的变异,这些贝勒流程不能筛除的变异可能在中国人群的变异频率比较高,而对于贝勒实验室内部数据库则为罕见变异或未检测出变异。本文有523个候选变异为复旦流程检出,而贝勒流程未检出,也与使用不同的内部数据库相关,内部数据库频率筛选的阈值贝勒流程为2%,复旦流程设置为10%,是造成这种差异的主要原因。
通过复旦流程的注释和筛选,可以在数分钟内将由WES检测到的70 000~100 000个变异缩减到仅需进行人工判读的100~200个变异,经验丰富的临床信息分析人员依据详细的基因功能、遗传模式、所致疾病、变异是否影响蛋白质结构预测信息等相关注释,可在较短的时间内锁定致病突变,完成对样本的分子遗传学诊断。
[1]Beaulieu CL, Majewski J, Schwartzentruber J, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet, 2014,94(6):809-817
[2]Firth HV, Wright CF, DDD Study. The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol, 2011, 53(8):702-703
[3]The Deciphering Developmental Disorders (DDD) study. http://www.ddduk.org/. Accessed 25/08/2014
[4]Tifft CJ, Adams DR. The National Institutes of Health undiagnosed diseases program. Curr Opin Pediatr, 2014, 26(6):626-633
[5]Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med, 2013, 369(16):1502-1511
[6]Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA, 2014, 312(18):1870-1879
[7]Jacob HJ. Next-generation sequencing for clinical diagnostics. N Engl J Med, 2013, 369(16):1557-1558
[8]Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat, 2003, 21(6):577-581
[9]https://www.bcm.edu/news/molecular-and-human-genetics/whole-exome-sequence-takes-new-tech-to-clinic .Aavaible at 2005-02-01
(本文编辑:张崇凡)
Evaluation of diagnostic accuracy of the whole-exome data analysis pipeline of Children's Hospital of Fudan University
LIZi-xiu1,LIUBo1,YANGLin,WANGHui-jun,ZHOUWen-hao(1DepartmentofBiostatisticsandComputationalBiology,LifeScience,FudanUniversity,Shanghai200433;2CentralChinaAgriculturalUniversity,Wuhan430072;3Children'sHospitalofFudanUniversity,Shanghai201102;4ShanghaiKeyLaboratoryofBirthDefects,TheTranslationalMedicineCenterofChildrenDevelopmentandDiseaseofFudanUniversity,KeyLaboratoryofNeonatalDiseases,MinistryofHealth,Children'sHospitalofFudanUniversity,Shanghai201102;5Co-firstauthor)
ZHOU Wen-hao,E-mail:zwhchfu@126.com
ObjectiveTo build data analysis pipeline applied for the high-throughput sequencing data analysis process for the clinical diagnosis of genetic diseases.Methods87 WES data of multiple malformations cases from the biobank of neonatal birth defect of Children's Hospital of Fudan University (our hospital). Candidate mutation screening was performed using the pipeline of published whole-exome sequencing from Baylor in the USA (Baylor pipeline) as a reference standard. The same batch of data was then run using high-throughput sequencing data analysis and clinical diagnosis pipeline (Fudan pipeline). The results were then compared line by line using the chromosome location and the base position of the mutations as the identifiers.ResultsThe total number of the variants detected in 87 cases was 7 820 777. After the quality control, selective filtering from public database, in-house database, and screening from mutation type and Mendelian diseases associated variance filtering, an average of 134-210 variation was as the final candidate for clinical diagnosis analysis. 15 242 candidate variations were filtered from Baylor pipeline, and 15 660 from Fudan pipeline, both of them hit 15 137 variations. 523 more variations were found by Fudan pipeline, but there were still 105 variants missed, which hit in Baylor pipeline. When compared with Baylor pipeline, the sensitivity, specificity, positive predictive percentage and negative predictive percentage were all higher than 96.66%.ConclusionFudan pipeline could simulate Baylor pipeline for the performing of WES data filtering, effectively acquire candidate variation. Though the current Fudan pipeline is only a basic framework, it provides an indispensable process of using WES in clinical practice.
Whole-exome sequencing; Neonates; Sensitivity; Specificity; Screening pipeline; Multiple malformations
上海市卫生局重要疾病攻关项目:2013ZYJB0015;上海市科委/医学领域重点项目子课题:14411950402,14DJ1400103;上海市卫计委项目:沪卫计科教〔2013〕018号
1 复旦大学生物统计学与计算生物学系 上海,200433;2 华中农业大学 武汉,430072;3 复旦大学附属儿科医院 上海,201102;4 上海市出生缺陷防治重点实验室,复旦大学儿童发育与疾病转化医学研究中心,卫生部新生儿疾病重点实验室,复旦大学附属儿科医院 上海,201102;5 共同第一作者
周文浩,E-mail:zwhchfu@126.com
10.3969/j.issn.1673-5501.2015.01.004
2015-01-03
2015-01-30)
