高通量测序数据分析和临床诊断流程的解读
2015-04-21黎籽秀徐凌丽王慧君周文浩
黎籽秀 刘 博 徐凌丽 杨 琳 王慧君 周文浩
·讲座·
高通量测序数据分析和临床诊断流程的解读
黎籽秀1刘 博2徐凌丽3杨 琳3王慧君4周文浩4
高通量测序技术也称二代测序技术,可以一次对几十至几百万条短序列片段同时进行测定。该技术的出现使研究者可以对一个物种的基因组、转录组和表观遗传组进行全面的分析。基因组二代测序可用于对全基因组、全外显子组或感兴趣的特定区域进行序列测定。通常,基因组二代测序的目的是检测个体基因组范围内的遗传变异,包括单碱基变异(SNVs)、插入缺失变异(Indels)、拷贝数变异(CNVs)和结构变异(SVs),并最终筛选出致病突变[1]。
近年来,基因组二代测序开始逐步应用于临床分子诊断[2~8],不仅能帮助医生明确患者的遗传学病因,指导治疗及判断预后,更重要的是可为遗传咨询提供明确的指导。根据患儿及其父母的遗传信息,可判断患儿致病成因是新生突变还是遗传于父母,以此评估父母生育下一胎时的疾病遗传风险,对家庭的“优生”提供更好的指导。
基因组二代测序技术与传统分子检测技术不同,可以同时对大量基因进行检测,一次性获得海量的数据。因此,构建一个基于遗传性疾病诊断需要的基因组二代测序数据分析流程,以期从众多变异中筛选出潜在致病突变,显得尤为重要。复旦大学附属儿科医院转化医学中心团队在美国贝勒医学院人类与分子遗传系的学习、交流和指导下,通过参阅既往数据分析相关文献[1,2,9]以及建立流程的实际经验,建立一套高通量测序数据分析和临床诊断流程(图1),包括测序数据预处理及变异检测、变异注释、变异筛选和变异分类等,清晰地向临床医生展现了变异筛选过程的概况,使研究者聚焦到更具有生物学意义、临床相关的变异,并为国内开展基于基因组二代测序技术的遗传性疾病诊断思考提供了基本路线图。复旦大学附属儿科医院转化医学中心应用该流程分析了87例多发畸形患儿的WES数据,得到的候选变异经由遗传专科医生进行分析,检出的阳性率为25%,与目前认为WES的检出阳性率一致。

图1 高通量测序数据分析和临床诊断流程
1 测序数据预处理及变异检测
基因组二代测序初始数据是由荧光或电信号组成的图像信息,图像信息可通过相应测序平台提供的软件经碱基识别(Base Calling)转化成FASTQ或FASTA格式的原始序列数据(Raw data)。Raw data去除接头以及低质量的读序后,采用BWA软件[10]将其定位到人类基因组的参考序列上,通过picard
(http://picard.sourceforge.net)和SAMtools软件[10]将建库过程中由于PCR扩增产生的冗余信息去掉。最后用GATK[11,12]检测变异,包括SNVs和Indels。目前,测序数据预处理及变异检测已形成较为成熟的生物信息分析流程。大多数测序公司均能提供完成此流程的服务。
2 注释
基因组二代测序技术产生了大量的遗传变异数据,其中仅少数变异具有功能意义。为了从众多变异中锁定可能的致病突变,需要从不同层面对变异进行注释。注释过程主要通过ANNOVAR和VEP(Variant Effect Predictor)软件及自行添加进行注释。
ANNOVAR[13]是第一个对遗传变异进行注释的软件。经过ANNOVAR 的注释,可对变异有多层面的了解,便于对其进行后续筛选。ANNOVAR对变异的注释包括以下3个方面,①基因的注释:注释信息包括变异类型,引起蛋白质一级结构改变的情况等。可以灵活地使用RefSeq基因、UCSC基因、ENSEMBL基因、GENCODE基因或其他基因定义系统进行位点-基因定位注释;②区域的注释:对变异位点所处的基因组环境进行注释,位点的基因组环境包括位点的保守性、转录因子、非转录RNA结合强弱和表观遗传标记物靶向性等信息;③过滤的注释:其注释结果可对变异进行后续筛选,包括变异在不同群体频率的注释,变异位点在dbSNP的注释,位点对蛋白质三维结构影响预测的注释,位点与疾病关联的注释。
VEP[14]是Ensembel和Ensembel基因组最常用的工具之一,用以研究变异对基因、转录本、蛋白质和调控区域所造成的影响。
虽同为变异注释软件,但VEP与ANNOVAR存在区别,①ANNOVAR选用NCBI的RefSeq参考序列注释,VEP选用Ensembl的转录本集合作为参考序列注释;②注释策略存在差异,ANNOVAR注释为同义突变的位点,VEP可能将其注释为错义突变。因此,2个注释软件针对同一个变异的注释结果可进行相互补充。
除使用上述2个软件进行注释外,还有诸如蛋白质序列数据库(Swiss-Prot)、人类基因突变数据库(HGMD)以及内部数据库(in-house database)等提供的重要参考信息需要人工添加进行注释。
2.1 基因注释参考的数据库
2.1.1 The Reference Sequence(RefSeq) http://www.ncbi.nlm.nih.gov/refseq/。RefSeq是美国国家生物信息技术中心(NCBI)提供的具有生物学意义的非冗余的DNA、RNA和蛋白质参考序列数据库。RefSeq为基因注释,突变及多态性分析,基因表达研究等提供了重要的参考标准[15]。RefSeq提供了基因的染色体号、基因所在染色体位置、基因转录起始终止位点、翻译起始终止位点和各个外显子的起始终止位置等信息。使用RefSeq对变异位点进行基因注释可明确发生变异的基因,变异所处基因功能区域,变异类型以及氨基酸改变的情况。
2.1.2 蛋白质序列数据(Swiss-Prot) http://www.uniprot.org/。Swiss-Prot是一个人工注释的、非冗余的蛋白质序列数据库。该数据库中的所有条目均由分子生物学家和蛋白质化学家通过计算机工具预测并查阅相关文献进行仔细核实。Swiss-Prot数据库是目前最全面的注释蛋白质序列库,其目的是对蛋白质提供全面的已知相关信息。许多序列分析软件被用于Swiss-Prot条目注释。软件分析结果通过人工评估后,被选择性地加入条目注释中。数据库中每个条目均有详细的注释,包括蛋白质、基因名字、蛋白质功能、表达模式、结构域、功能位点、跨膜区域、二硫键位置、翻译后修饰、突变体及其与疾病的关系等。Swiss-Prot目前包含547 357例条目,并与其他30多个数据库交叉引用,例如PDB、OMIM和PROSTITE等。
2.2 变异/基因与疾病关系注释参考的数据库
2.2.1 人类基因突变数据库(HGMD)[16]http://www.hgmd.cf.ac.uk/ac/index.php。HGMD由英国卡尔地夫医学遗传研究所构建。HGMD用计算机和手工结合的方法从已发表在期刊中收集与人类遗传疾病相关的突变信息,是目前收录人类突变信息最全的数据库。截至2014年12月18日,HGMD收录疾病相关变异的数量在免费版本中为108 508个,在专业版本中为156 932个[17]。HGMD中记录的突变信息包括突变类型列表、对应的疾病列表和相应的参考文献。其中突变类型包括在编码区、调控区和剪接区域中的大片段插入缺失、微片段插入缺失、基因组重组、重复变异、致病点突变、致病点移码、致病点无义、影响可变剪接的变异和与疾病相关的多态性位点,包括在所研究的疾病组和对照组差异有统计学意义的位点和已被报道能影响基因表达或蛋白质结构和功能的位点。HGMD的免费版本只能在线查找基因变异信息[16]。
HGMD根据变异位点和疾病的关联程度以及位点的突变类型等信息对位点进行分类,包括①致病突变(DM),即目前认为该突变能直接导致疾病;②疑似致病突变(DM?),即该变异曾被认为是致病突变,但基于基因组/群体筛查或其他发现该突变可能与病理无关或为中性突变。随着突变信息的累积,若该变异被确认不是DM,可能会被彻底从数据库删除; ③疾病相关的多态性变异(DP),即疾病/表型显著相关的多态性位点,根据位点复制、进化保守性等信息认为这些变异有一定的功能,但目前尚无功能实验证实(如表达研究);④有功能证据支持的疾病相关多态性变异(DFP),即有功能实验证实(如:表达结果改变,mRNA研究等)的疾病/表型显著相关的多态性位点;⑤体外/实验室或体内功能的多态性变异(FP),即影响一个基因(或基因产物)的结构、功能或表达,但目前尚未报道与疾病关联的多态性位点;⑥移码或truncating变异(FTV),即被预测能引起基因编码蛋白质的改变或截短,但目前未报道与疾病关联或致病的变异。
DP、DFP、FP和FTV类型的变异约占HGMD报道变异的5.5%,同时,这4种类型的变异直接致病性证据不强,所以WES流程筛选罕见遗传性疾病的致病突变时应当优先关注DM和DM?。
2.2.2 在线人类孟德尔遗传数据库(OMIM)[18]http://omim.org/。OMIM是由美国约翰霍普金斯大学于1968年建立的关于人类基因和基因突变的数据库。每日更新以提供全面而权威的基因遗传和疾病表型信息,截至2014年12月15日,OMIM数据库收录了22 700个词条;分子基础已知的表型有5 369个,疾病基因有3 309个。OMIM条目包括基因和突变的文字描述、病例记录、分子诊断、参考文献和与其他数据库的链接。
区别于HGMD提供基因所有的变异位点信息,大多数OMIM基因更关注疾病基因的第一个突变,对应表型最常见的突变以及具有不寻常特征的突变,包括特殊突变类型,特殊突变致病机制,特殊突变遗传模式(如在相同基因中,部分突变为显性遗传模式,部分为隐性遗传模式)等。除此之外,OMIM提供较为全面的疾病临床表型谱,为临床医生和研究者根据患者的表型信息对诊断疾病提供依据[19]。
OMIM数据库存储的疾病信息以孟德尔遗传病为主,近年来也收录了许多复杂疾病以及复杂疾病易感的多态性位点等信息[20]。因此,在研究罕见遗传疾病时,需要对OMIM数据库中的疾病分等级对待,具有OMIM号的单基因疾病优先被考虑。
2.2.3 Clinvar[21]http://www.ncbi.nlm.nih.gov/clinvar/。为了促进和加速对基因型与表型之间关系的研究,NCBI于2013年4月正式启动ClinVar公共免费数据库。ClinVar数据库旨在整合NCBI以及各种遗传变异和临床表型数据库,通过标准的命名法来描述疾病,将变异、临床表型、实证数据和功能注解与分析4个方面的信息,通过专家评审,逐步形成一个标准的、可信和稳定的遗传变异-临床表型相关的数据库。
ClinVar数据库与其他“变异/基因-疾病”数据库的重要区别在于该数据库有一系列的专家小组对大量数据进行评估和归纳,能更好地理解基因型和重要表型之间的关系。对于感兴趣的变异,ClinVar除了列举突变类型,突变与疾病对应关系等基本信息外,还包括该变异的临床意义(分为9个类别,其中类别4和5在寻找罕见遗传性疾病候选突变时应优先考虑),专家对变异与疾病关联可信度的评价(分为4星级,变异和疾病的关联可信度达到3星级以上则表明该变异已通过专家小组的评估审核,可明确变异和疾病存在关联性)。2.3 突变频率注释参考的数据库
2.3.1 千人基因组计划(1000 Genome Project) http://www.1000genomes.org/ 。“千人基因组计划”是2008年初由英国Sanger研究所、美国国立人类基因组和中国华大基因研究所共同启动的、以二代测序技术为主导的人类基因组计划三期工程。千人基因组的数据发现,每个人平均携带250~300个未报道过的变异,其中50~100个变异与遗传病有关[22]。千人基因组计划项目的开展,不仅加速了对常见疾病易感性基因的发现,还将加深对人类基因组结构差异的认识,为解释人类重大疾病的发病机制,开展疾病个性化预测、预防和治疗奠定了基础。千人基因组计划完成了基因组科学从基础向应用过渡的关键战略转移,有效地推进了临床转化医学的兴起和发展[23]。
2.3.2 The Exome Aggregation Consortium(ExAC) http://exac.broadinstitute.org/。ExAC是一个专门研究外显子组测序数据的联盟机构,整合了多个外显子组测序计划。截止于2014年12月3日,数据库收录了91 796个样本的外显子测序数据,其中包括61 486个独立样本的数据。为了更好地统计变异频率,ExAC使用相同的测序数据预处理及变异检测分析流程对外显子测序数据进行处理,即以GRCH37/hg19基因组作为人类基因组参考序列,用dbSNP135对变异进行注释。ExAC是目前收录不包含严重儿童疾病样本的最大数据库,因此该数据库能更好地作为研究儿童孟德尔遗传病的对照[24]。
2.4 变异预测注释参考的软件
2.4.1 SIFT(Sorting Intolerant From Tolerant)[25,26]http://sift.jcvi.org/。SIFT是一种基于序列同源性对氨基酸的替换容忍度进行评分,以预测氨基酸替换是否影响表型的软件。2001年,Ng和Henikoff发现重要的氨基酸位点在蛋白质家族序列中较为保守,这些保守位点上发生的氨基酸替换更有可能影响蛋白质功能。基于该假设,Ng和Henikoff采用位置相关评分矩阵(PSSM)来描述序列保守性信息[27],开发了预测错义突变对蛋白质功能影响的软件SIFT。SIFT分数归一化后范围为0~1,其中,分数<0.05是有害替换(Deleterious),≥0.05是可容忍的替换(Tolerate)。值得注意的是,应用SIFT软件对错义突变进行功能预测的前提是必须有足够的同源序列,否则其预测精度将下降,甚至无法进行预测[28]。
2.4.2 Polyphen-2 (Polymorphism Phenotyping v2)[29]http://genetics.bwh.harvard.edu/pph2/。Polyphen-2是通过整合蛋白质序列和蛋白质三维结构特征,来预测人类蛋白质的氨基酸替换对结构和功能影响的软件。采用贪婪迭代算法,从19个基于序列和13个基于结构的特征中,自动选取了8个基于序列和3个基于结构的特征来进行预测。其中序列特征包括变异位点所处于在蛋白质结构域(Pfam)的位置信息,是否导致CpG位点发生转换(Transition)等,蛋白质结构特征包括溶剂可及性、SNP位点在 β链或活动区域的位置等。该方法有较高敏感度和特异度的前提是有可靠的蛋白质结构信息进行参考。Pholyphen-2有HumVar和HumDiv两种模型。在对于孟德尔遗传病的诊断分析中,HumVar模型产生的分数更适用于诊断。运行Pholyphen-2算法进行打分后,分数的范围为0~1。分数越高的替换意味着有越大的破坏蛋白功能的可能,如果分数在0.957~1,其相应的预测结果为“probably damage”,在0.453~0.956为“possible damage”,在0~0.452为“benign”。
2.4.3 MutationTaster[30]http://www.mutation-taster.org/。MutationTaster是通过使用进化保守性、剪切位点改变和mRNA水平的变化引起的蛋白质特征丢失等信息,来评估序列变异带来的致病可能性的软件。HGMD专业版本中提供的390 000个已知致病突变位点信息作为阳性数据集,千人基因组计划中>6 800 000个无致病突变的多态性信息作为阴性数据集,用贝叶斯分类算法对阴、阳性数据集建模,对感兴趣的位点进行预测,预测结果的分数为0~1,分数越高意味着致病可能性越大,根据预测提示的分数及先验信息校正后,软件会对变异的致病可能性进行分类,具体说明如下:①A:disease_causing_automatic,变异在ClinVar中标记为致病性或者该变异是导致终止密码子提前的无义突变;②D: disease_causing,变异被软件预测为致病性突变;③N: polymorphism,变异被软件预测为多态性;④P: polymorphism_automatic,变异在HapMap数据中存在3种基因型 AA、AB和BB或在千人基因组计划数据集中显示纯合突变频数>4。
3 筛选
3.1 质量控制 针对某个特定位点,若覆盖该位点的读序总数小于覆盖该位点变异和未变异碱基的读序数目之和,则表明该位点的质量未达标,应将其去除。该筛选过程可将小部分的SNVs和约一半的Indels筛除。
3.2 频率筛选 基因变异程度可根据最小等位基因频率(MAF)进行划分。MAF值5%~50%的变异为常见变异,1%~5%为少见变异,<1%则为罕见变异[31]。基于罕见疾病是由罕见变异所导致的这一假说,在研究罕见疾病的致病变异时,应去除非罕见变异。
基于变异频率的筛选方式为:① 变异已被HGMD报道,表明该变异更有可能与疾病相关,此时将注释的公共数据库的变异频率筛选阈值设置为5%。②变异未被HGMD报道,此时将注释的公共数据库的变异频率筛选阈值设置为1%[2]。③如果研究机构拥有收录不同疾病患者信息的内部数据库,若变异在内部数据库10%的家系中出现,则应当去除该变异;当内部数据库中无关个体数量达到1 000例时,筛选阈值可降至4%。使用内部数据库的优势在于:一方面内部数据库位点的变异频率更符合中国人群的变异频率,另一方面可去除由相同测序平台导致的系统误差。
3.3 分类筛选 根据所处基因组位置的不同,可将变异分为编码区变异和非编码区变异。研究发现,85%的致病突变都位于编码区中,其中绝大部分位于外显子上;极少数位于内含子上,可通过影响基因的可变剪接致病[32]。没有被HGMD报道的位于非编码区的变异,或者距离外显子区>5 bp的内含子变异(不能影响mRNA的剪接)被筛选掉[2]。
保留下来的突变,根据突变对蛋白质序列影响的不同,可以分为同义突变、错义突变、无义突变、终止密码突变、剪接位点突变、移码突变和整码突变。无义突变、剪接位点突变和移码突变被称为truncating突变,能造成蛋白质缺失。同义突变不会引起蛋白质一级结构的改变,整码突变与终止密码突变虽然一定程度上改变蛋白质一级结构,但由于保留了基因阅读框的次序,所以一般情况下不会造成蛋白质的功能缺陷。因此,寻找罕见疾病的候选致病突变优先考虑能引起蛋白质功能缺陷的变异。
4 变异分类
本文通过参阅ACMG(American College of Medical Genetics)[33]对变异的分类标准,并结合实际研究经验,基于基因是否在OMIM/HGMD中被报道为致病基因,同时考虑突变频率和类型,既往报道和临床表现,根据变异的临床可信度对变异进行分类。
4.1 已报道致病突变位点 已经被报道为致病突变,并且既往报道该变异导致疾病的表型谱和患者的临床表型相符。
4.2 新突变但预测为致病突变 包括无义突变、终止密码突变、起始密码子(ATG)突变、移码突变和剪接供体/受体突变。
4.3 新突变致病性不明确 包括剪接共有序列突变、错义突变和整码突变。
4.4 报道与临床表型相关联但致病性不明确 即通过全基因组关联分析(GWAS)得到的与复杂疾病易感性相关的变异。
4.5 其他 不满足上述4类的变异,如:尚不认为能导致疾病的新变异,新发的同义突变;已报道为中性突变的变异;在公共数据库中有一定突变频率的罕见变异,这类罕见变异部分可构成常染色体隐性遗传模式而致病。

5 结合遗传模式和临床综合判断
针对候选致病突变,应当进一步具体结合相关疾病的遗传模式以及患者实际的临床表型进行综合判断。
就单基因病的常染色体遗传模式而言,如果疾病为常染色体显性遗传模式,则其致病基因一般仅发生单个位点的严重突变,且该突变在正常人群中极有可能为新发突变。如果疾病为常染色体隐性遗传模式,则其致病基因上将发生至少2个严重突变。符合这种遗传模式的突变,可在正常人群中有一定的突变频率,但一般情况下不会出现纯合子[2]。若在致病基因上发生的突变不符合相关疾病的遗传模式(如疾病为常染色体隐性遗传模式,但在该疾病相关基因上仅发生单位点杂合突变),该突变位点应当被滞后考虑[38]。
对于患者的潜在致病突变,须将其临床表型与突变基因对应的表型谱进行比对。收集诸如患者病历、家族史等信息,将有助于明确患者的致病成因[1]。
[1]Bao R, Huang L, Andrade J, et al. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform, 2014, 13(Suppl 2):67-82
[2]Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med, 2013, 369(16):1502-1511
[3]Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA, 2014, 312(18):1880-1887
[4]Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA, 2014, 311(10):1035-1045
[5]Berg JS. Genome-scale sequencing in clinical care: establishing molecular diagnoses and measuring value. JAMA, 2014, 312(18):1865-1867
[6]Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet, 2012, 49(6):353-361
[7]Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA, 2014, 312(18):1870-1879
[8]Eng CM, Yang Y, Plon SE. Genetic diagnosis through whole-exome sequencing. N Engl J Med, 2014, 370(11):1068
[9]Wu L, Schaid DJ, Sicotte H, et al. Case-only exome sequencing and complex disease susceptibility gene discovery: study design considerations. J Med Genet, 2015, 52(1):10-16
[10]Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009, 25(16):2078-2079
[11]McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res, 2010, 20(9):1297-1303
[12]Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics, 2013, 11(1110):11.10.1-11.10.33
[13]Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 2010, 38(16):e164
[14]McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics, 2010, 26(16):2069-2070
[15]Maglott DR, Katz KS, Sicotte H, et al. NCBI′s LocusLink and RefSeq. Nucleic Acids Res, 2000, 28(1):126-128
[16]Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat, 2003, 21(6):577-581
[17]Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet, 2014, 133(1):1-9
[18]Schorderet DF. Using OMIM (On-line Mendelian Inheritance in Man) as an expert system in medical genetics. Am J Med Genet, 1991, 39(3):278-284
[19]Zhuang YL(庄永龙), Zhou M, Li YD, et al. The Application of Human Mutation Databases. Hereditas(Beijing)(遗传), 2004, 26(4):514-518
[20]Amberger JS, Bocchini CA, Schiettecatte F, et al. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res, 2015, 43(Database issue):789-798
[21]Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014, 42(Database issue):980-985
[22]1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, et al.A map of human genome variation from population-scale sequencing.Nature, 2010,467(7319):1061-1073
[23]1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature, 2010, 467(7319):1061-1073
[24]http://macarthurlab.org/2014/11/18/a-guide-to-the-exome-aggregation-consortium-exac-data-set/
[25]Sim NL, Kumar P, Hu J, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res, 2012, 40(Web Server issue):452-457
[26]Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res, 2001, 11(5):863-874
[27]Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet, 2006, 7:61-80
[28]Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc, 2009, 4(7):1073-1081
[29]Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods, 2010, 7(4):248-249
[30]Schwarz JM, Rödelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods, 2010, 7(8):575-576
[31]Sui WG(眭维国), Li LP, Che WT, et al. 人类遗传疾病中常见变异和罕见变异的研究策略. Int J Lab Med(国际检验医学杂志), 2011, 32(16):1847-1850
[32]Robinson PN, Krawitz P, Mundlos S. Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin Genet, 2011, 80(2):127-132
[33]Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med, 2008, 10(4):294-300
[34]Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet, 2010, 42(9):790-793
[35]Robinson PN, Köhler S, Oellrich A, et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res, 2014, 24(2):340-348
[36]Pippucci T, Parmeggiani A, Palombo F, et al. A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One, 2013, 8(12):e82154
[37]Smedley D, Köhler S, Czeschik JC, et al. Walking the interactome for candidate prioritization in exome sequencing studies of Mendelian diseases. Bioinformatics, 2014, 30(22):3215-3222
[38]Zemojtel T, Köhler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med, 2014, 6(252):252ra123
(本文编辑:张崇凡)
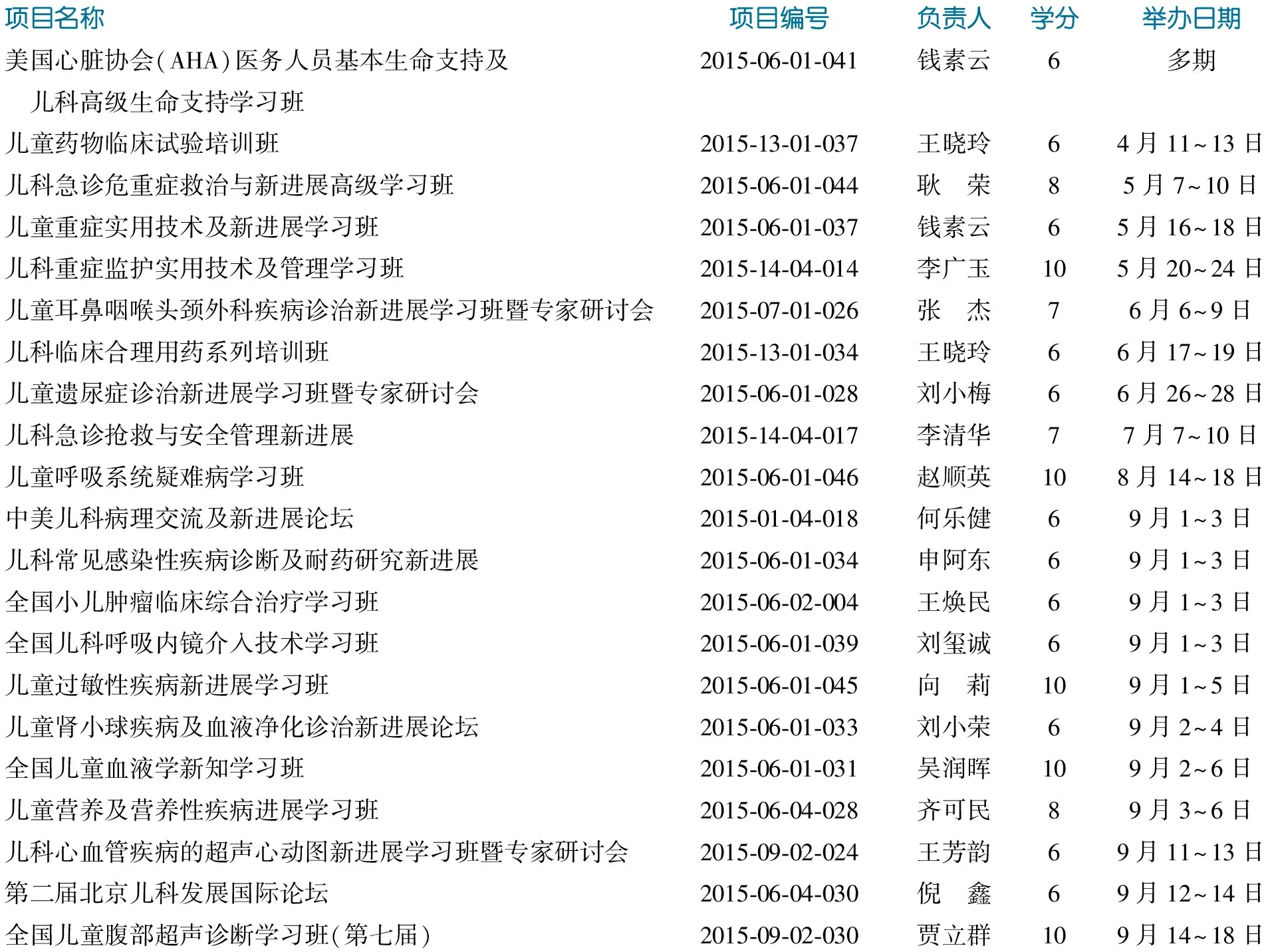
首都医科大学附属北京儿童医院2015年国家级继续医学教育项目(一)
10.3969/j.issn.1673-5501.2015.01.003
上海市卫生局重要疾病攻关项目:2013ZYJB0015;上海市科委/医学领域重点项目子课题:14411950402,14DJ1400103;上海市卫计委项目:沪卫计科教〔2013〕018号
1 复旦大学生物统计学与计算生物学系 上海,200433;2 华中农业大学 武汉,430072;3 复旦大学附属儿科医院 上海,201102;4 上海市出生缺陷防治重点实验室,复旦大学儿童发育与疾病转化医学研究中心,卫生部新生儿疾病重点实验室,复旦大学附属儿科医院 上海,201102
周文浩,E-mail:zwhchfu@126.com
2014-12-17
2015-01-20)
