头孢氨苄不稳定性对建立相应复方制剂质量标准的影响
2015-03-11韩宁宁于丽娜徐嫄郝利华赵晖
韩宁宁,于丽娜,徐嫄,郝利华,赵晖
(中国兽医药品监察所,北京100081)
头孢氨苄不稳定性对建立相应复方制剂质量标准的影响
韩宁宁,于丽娜,徐嫄,郝利华,赵晖*
(中国兽医药品监察所,北京100081)
在兽用复方化学制剂头孢氨苄单硫酸卡那霉素乳房注入剂的注册检验过程中发现:当萃取所用的乙醚为国产分析纯试剂时,头孢氨苄会降解从而影响相应含量测定与降解物检查;在卡那霉素降解物检查项中,供试品溶液制备过程中的加热步骤会导致头孢氨苄降解从而干扰该项测定。针对上述两个问题进行研究后认为,申报方在标准中应对试剂纯度及卡那霉素降解物检查项中空白的配制方法作出详细规定。本实验在为完善该质量标准提供参考的同时,也可为同业人员制定含有不稳定主成分的兽用复方化学制剂的质量标准提供借鉴。
头孢氨苄;单硫酸卡那霉素;试剂纯度;不稳定性
复方制剂由两种或两种以上药物组成,在药品注册申请中已占有一定的比例,与单方制剂相比,其质量研究工作更复杂,存在的问题也更多样化。处方内主成分的不稳定性、主成分间的相互作用对杂质检查的影响都应在相应标准制定过程中给予关注。
近日,我们对某申报进口注册的头孢氨苄单硫酸卡那霉素乳房注入剂进行了注册检验。该产品为复方制剂,其中头孢氨苄属第一代头孢菌素,卡那霉素属氨基糖苷类抗生素,二者合用,可提高后者在菌体内的浓度,进而呈现协同作用[1]。然而,头孢氨苄稳定性差,遇酸遇热均易分解[2],这一特性对该复方制剂的含量测定及有关物质检查均会产生影响,需要在质量标准建立过程中加以注意。我们在对兽用复方化学制剂头孢氨苄单硫酸卡那霉素乳房注入剂的注册检验过程中也发现了这样的问题并进行了研究。
1 材料与试药
高效液相色谱仪(HPLC),配紫外检测器(UV)和二极管阵列检测器(PDA)(Waters 2695-2487/996);AX-205分析天平(Mettler Toledo)。分析纯石油醚和乙醚购于国药集团;色谱纯乙醚和水购于Honeywell公司;液相用水为MILLI Q纯水机过滤的纯化水。头孢氨苄对照品、卡那霉素对照品及7-氨基3-去乙酰氧基头孢烷酸(7-ADCA)对照品均由申报方提供。头孢氨苄对照品批号20676,含量93.4%;卡那霉素对照品批号20889,含量779.1单位/mg;7-ADCA对照品批号WS00019,含量99.3%。供试品头孢氨苄单硫酸卡那霉素乳房注入剂规格:10g:头孢氨苄0.2g+卡那霉素10万单位。
2 方法与结果
2.1 不同纯度级别的试剂对头孢氨苄含量测定及头孢氨苄降解物检查的影响
2.1.1 申报方质量标准的相关规定
2.1.1.1 【含量测定】头孢氨苄 色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;以0.01 mol/L 乙酸钠溶液-乙腈-甲醇(910∶86.4∶3.6)为流动相;流速为1.5 mL/min;检测波长为260 nm;理论板数按头孢氨苄峰计算不低于1500。
测定法:精密称取本品约0.5 g,置分液漏斗中,加乙醚25 mL和石油醚25 mL,摇匀,用水提取3次,每次25 mL,合并水液至100 mL量瓶中,用水稀释至刻度,摇匀,取25 uL注入液相色谱仪,记录色谱图;另取头孢氨苄对照品适量,精密称定,用水溶解并稀释制成每1 mL中含头孢氨苄0.1 mg的溶液,同法测定。按外标法以峰面积计算供试品中头孢氨苄的含量。
2.1.1.2 【检查】7-ADCA和其他未知杂质(头孢氨苄降解物) 照头孢氨苄含量测定项下的方法测定,记录色谱图至主成分峰保留时间的1.5倍。供试品溶液色谱图中如有杂质峰,按下式分别计算7-ADCA、单个未知杂质含量和杂质总量,7-ADCA不得大于1.0%,单个未知杂质不得大于1.0%,杂质总量不得大于2.5%。
7-ADCA含量(%)=(7-ADCA峰面积/头孢氨苄峰面积)×P×0.7
单个未知杂质含量(%)=(未知杂质峰面积/头孢氨苄峰面积)×P×1
杂质总量(%)=7-ADCA(%)+未知杂质(%)+D-苯甘氨酸(%)
式中:0.7为7-ADCA的响应因子;1为未知杂质的响应因子;P为头孢氨苄的百分含量。
2.1.2 不同纯度级别试剂影响的比较 在实验初期发现,头孢氨苄含量测定结果偏低(结果为约75%),且供试品溶液色谱图中保留时间约2.5 min及9.1 min附近有两个较大的杂质峰(图1)超出了头孢氨苄降解物项的规定。由于实验初期萃取的溶剂均采用国药集团化学试剂有限公司生产的分析纯试剂及MILLI Q纯水机过滤的纯化水,我们考虑是否有可能受到溶剂的影响导致头孢氨苄的降解。
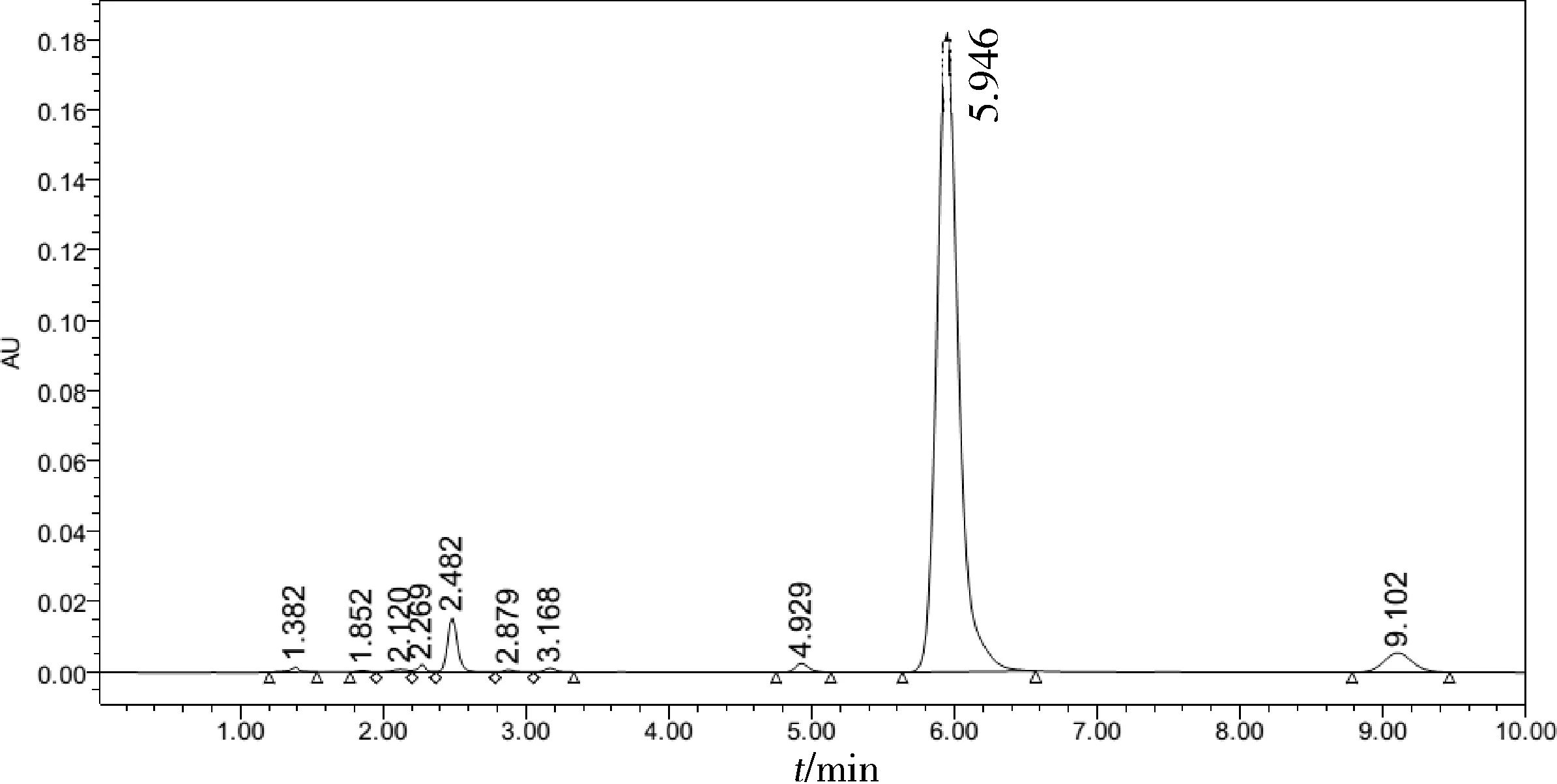
图1 使用分析纯乙醚和MILLI Q纯水机过滤的水萃取的头孢氨苄含量测定供试品溶液色谱图
由于查询到没有色谱纯石油醚,因此我们将剩余的两种溶剂——乙醚和水全部更换为Honeywell公司生产的色谱纯乙醚和水,结果发现头孢氨苄含量和降解物项均符合规定(图2)。
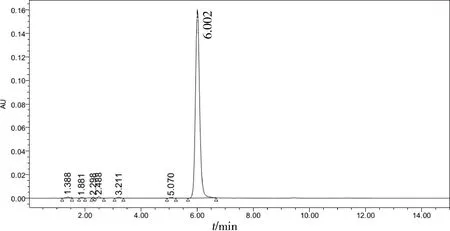
图2 使用色谱纯乙醚和水萃取的头孢氨苄含量测定供试品溶液色谱图
然后又将色谱纯乙醚更换回分析纯乙醚,或者将色谱纯水更换回MILLI Q纯水机过滤的纯化水,结果发现前者仍存在含量低和降解物不符合规定的问题,而后者两项结果均符合规定(表1)。至此,我们可以确定,国产分析纯乙醚会导致该申报制剂中头孢氨苄降解,进而影响测定结果。这可能是由于该分析纯乙醚中的杂质或者其偏酸性的pH值导致的。因此我们建议,申报方在标准中注明对萃取溶剂的要求。

表1 不同纯度级别的试剂对头孢氨苄含量测定及头孢氨苄降解物检查的影响
2.2 头孢氨苄的不稳定性对卡那霉素降解物检查的影响
2.2.1 申报方质量标准的相关规定
2.2.1.1 【含量测定】卡那霉素 色谱条件与系统适用性试验用辛基硅烷键合硅胶为填充剂(Phenomenex Luna C8,5 μm,250 mm×4.6 mm色谱柱);以水-乙腈(45:55)为流动相;流速为1.5 mL/min;检测波长为350 nm;理论板数按卡那霉素峰计算不低于1500。
测定法:精密称取本品约3.8 g,置250 mL分液漏斗中,加庚烷50 mL使主成分溶解,再用水提取3次,每次25 mL,合并水液至100 mL量瓶中,用0.4 mol/L三羟甲基氨基甲烷溶液溶解并稀释至刻度,摇匀;精密量取5 mL,置50 mL量瓶中,加0.15 mol/L 2,4-二硝基氟苯的甲醇溶液15 mL,在60 ℃水浴中加热60 min,冷却至室温,用50%乙腈溶液稀释至刻度,摇匀。精密量取10 uL注入液相色谱仪,记录色谱图;另取单硫酸卡那霉素对照品约50 mg,精密称定,置100 mL量瓶中,用0.1 mol/L三羟甲基氨基甲烷溶液溶解并稀释至刻度,摇匀;精密量取5 mL,置50 mL量瓶中,同法测定。按外标法以峰面积计算供试品中卡那霉素的含量。
2.2.1.2 【检查】卡那霉素降解物 照卡那霉素含量测定项下的方法测定,记录色谱图至主成分保留时间的1.5倍,供试品溶液色谱图中如有杂质峰,按下式计算单个杂质含量和杂质总量,单个杂质不得大于1.0%,杂质总量不得大于2.0%。
杂质含量(%)=杂质峰面积/卡那霉素峰面积×P
式中:P为卡那霉素的百分含量。
2.2.2 头孢氨苄不稳定性对空白色谱图的干扰 在实验中发现,不加供试品的空白色谱图中有多个色谱峰,可能是来源于萃取及稀释所用的溶剂或衍生化试剂(图3)。然而,将供试品图谱中的空白峰不计后,卡那霉素降解物项仍不符合规定(保留时间为9.3 min及10.2 min的两个杂质含量均大于1.0%)(图4)。考虑到该复方制剂中头孢氨苄热稳定性差,而卡那霉素降解物检查项中供试品溶液的制备过程,衍生化一步需要在60 ℃水浴中加热60 min,这一操作可能导致头孢氨苄降解,其产生的降解物峰有可能干扰卡那霉素降解物项的测定。
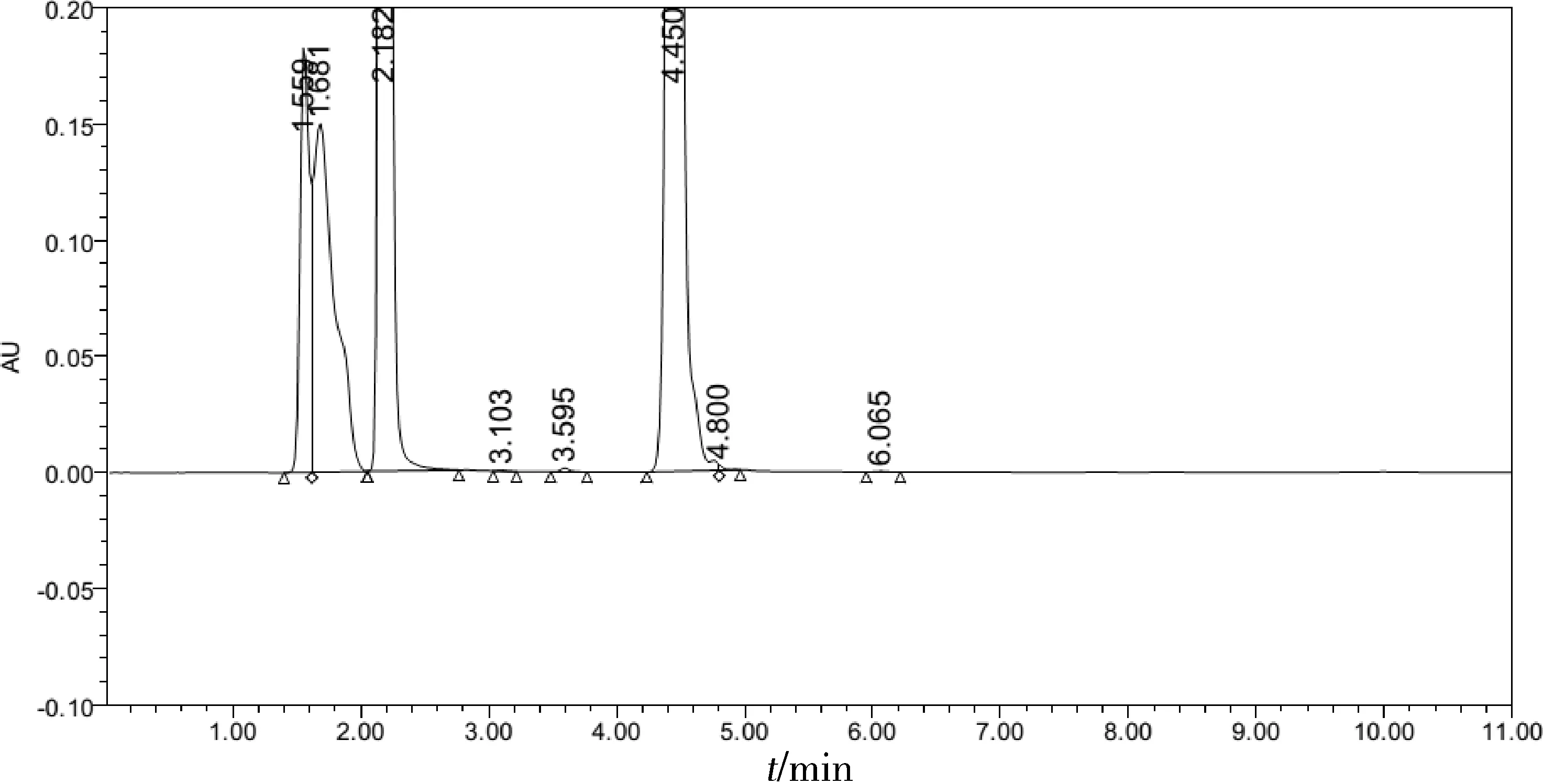
图3 卡那霉素降解物检查空白色谱图
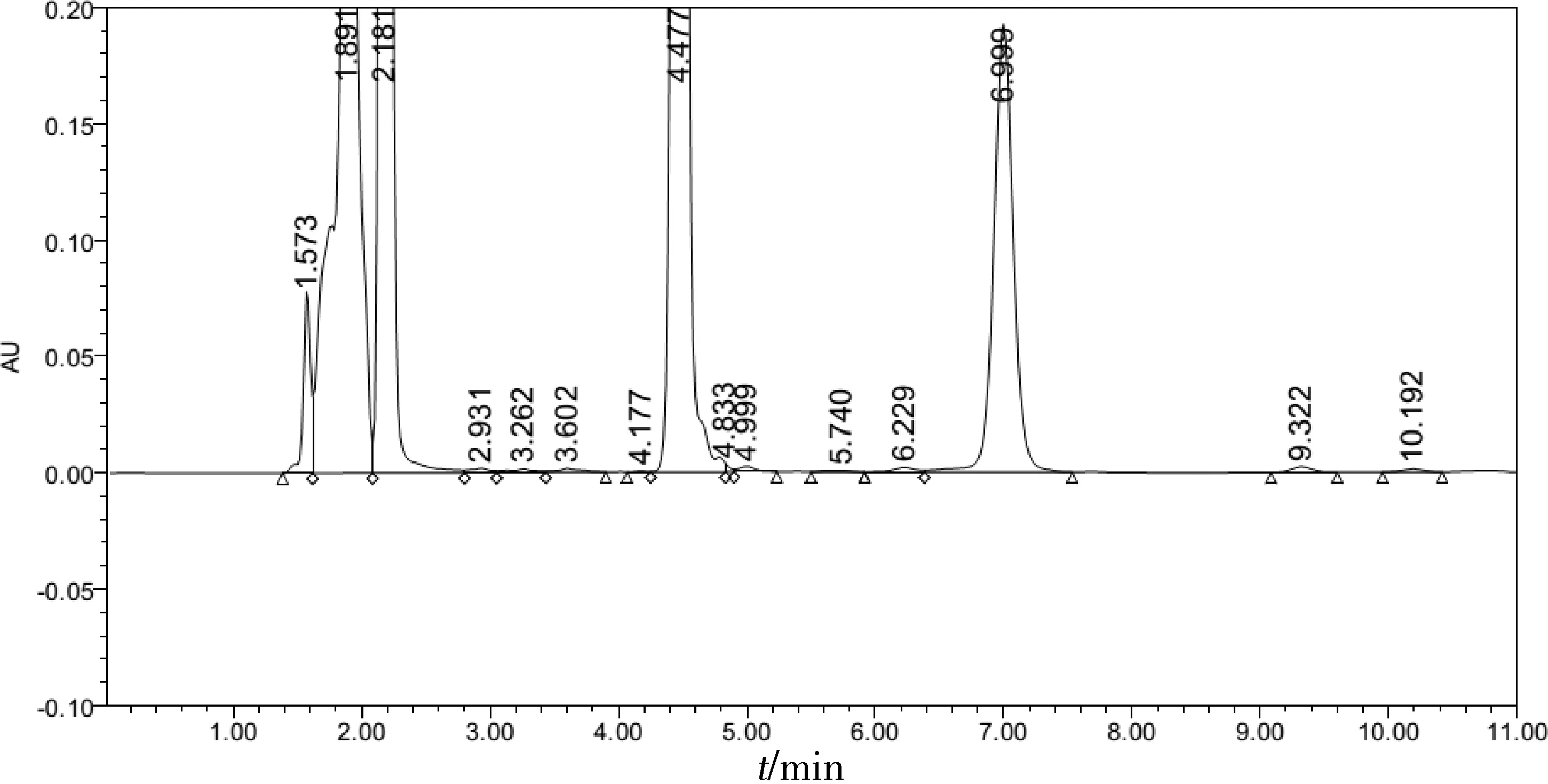
图4 卡那霉素降解物检查供试品溶液色谱图
因此,我们参照供试品中的头孢氨苄规格(10 g∶0.2 g),模拟配制了含有相应量的头孢氨苄(3.8 g×0.2 g/10 g)的空白溶液,并将其进行与供试品相同的处理。以此溶液的色谱图作为空白色谱图(图5)。将供试品图谱中的该空白峰不计后,卡那霉素降解物项符合规定。因此我们建议,申报方在标准中注明空白溶液的制备方法。

图5 添加头孢氨苄的卡那霉素降解物检查空白色谱图
3 讨论
申报进口注册的质量标准起草过程大多是在申报公司的所在国家完成,但是依据申报标准在我国进行注册检验时,试剂、水等条件往往不同。尤其对于像头孢氨苄这样极其不稳定的品种,不同纯度级别的试剂就可能对其测定产生影响。因此,申报方应尽可能对试验条件进行详细规定,以免产品本身合格而注册检验结果出现不符合规定的情况。
与单方制剂相比,复方制剂杂质检查项研究工作更为复杂,不仅要考虑每种标称成分的稳定性及辅料的影响,还要考虑标称成分之间相互作用的影响。练富林等[3]综述了化学药物复方制剂中杂质的检查与控制,其中总结了复方制剂中杂质的归属分析步骤(图6)。
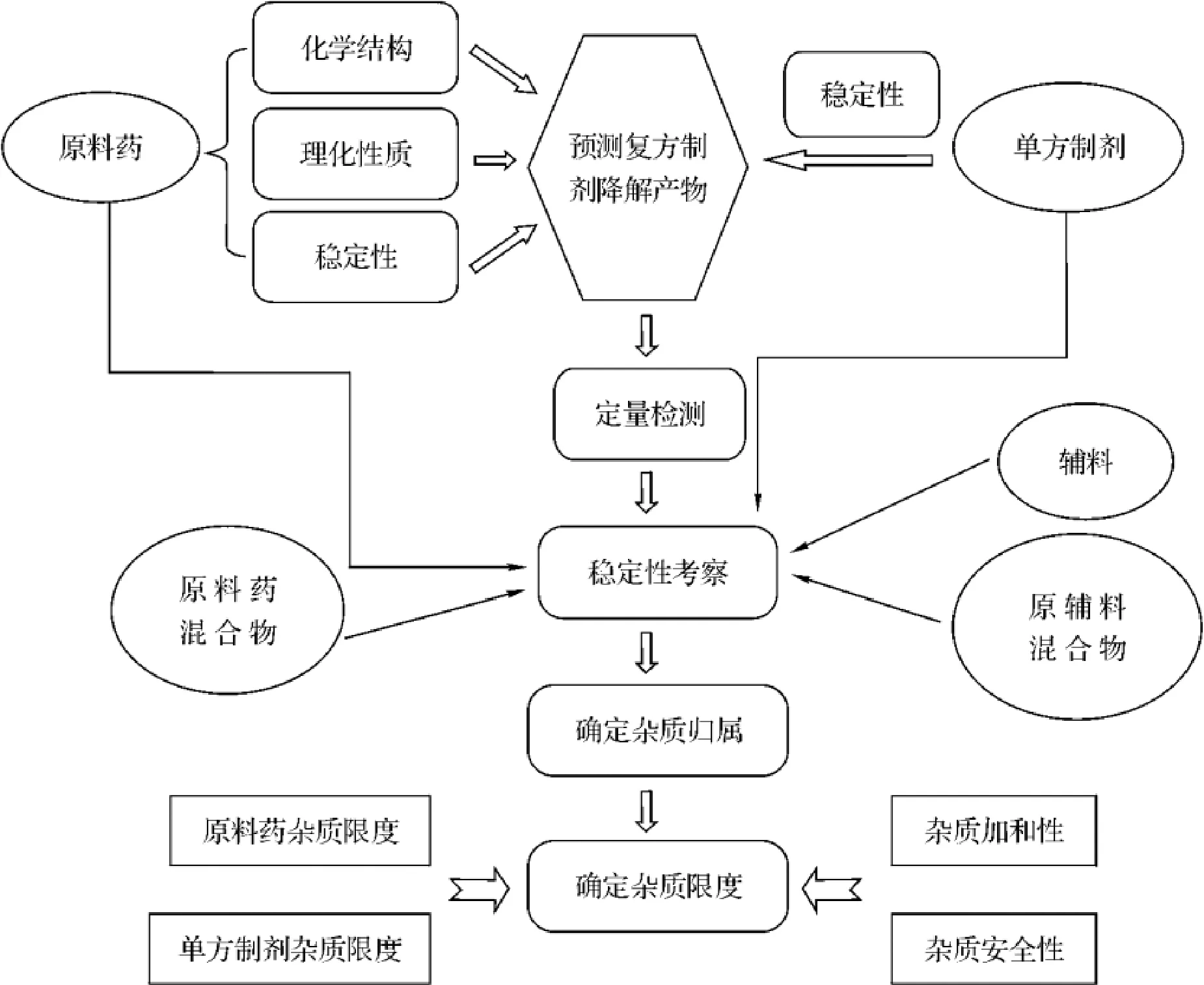
图6 复方制剂中杂质的归属分析步骤[3]
对于复方制剂中可能存在的杂质进行预测分析时,应首先通过查阅复方制剂中各原料药的相关文献,了解原料药中可能存在的杂质,从而推测复方制剂中的杂质;其次,对各原料药的化学结构、理化性质进行分析,并结合其生产工艺和制剂过程及贮藏条件,再推测可能因降解而产生的杂质。对复方制剂中可能存在的降解产物进行初步预测后,选择适宜的检测方法,保证各主药、杂质和辅料能得到有效分离、检出,并分别对各原料药及其混合物、辅料、原料与辅料混合物、制剂等进行稳定性考察,确定杂质来源及种类。
复方制剂中含量测定或有关物质检查项的前处理条件要充分考虑到所有标称成分的影响,既要使测定方法具有专属性,避免复方内另一种或另几种主成分的干扰,又要避免条件过于剧烈使测定成分本身降解。归根结底,还是要对复方制剂中所有原料药的化学结构、理化性质及稳定性有充分了解与考察,才能设计相对合理的测定方法。
[1] 中国兽药典委员会.中华人民共和国兽药典兽药使用指南化学药品卷[M].北京:中国农业出版社, 2010:31.
[2] 刘春萍,李贺一,吴志军.头孢氨苄片稳定性考察[J].黑龙江医药,2001,14(3):207.
[3] 练富林,李洪雪,施月芹, 等.化学药物复方制剂中杂质的检查与控制[J].药学进展,2009,33(3):112.
(编辑:李文平)
The Influence of the Instability of Cefalexin on Establishment of the Quality Standard of the Corresponding Compound Preparation
HAN Ning-ning, YU Li-na, XU Yuan, HAO Li-hua, ZHAO Hui*
(ChinaInstituteofVeterinaryDrugControl,Beijing100081,China)
During the registration test of veterinary compound preparation intramammary infusions of cephalexin and kanamycin monosulfate, two problems were found: when the ether used for extraction was analytical reagent, cephalexin degraded that would interfere with the content determination of cephalexin and the inspection of degradation of cephalexin; during the inspection of degradation of kanamycin, the heating process during the preparation of test sample solution could lead to degradation of cephalexin that would interfere with the determination.In view of the above two questions, the corresponding suggestions were put forward—the applicant should make detailed provisions on the purity of the reagent and the preparation method of the blank in the examination of the degradation of kanamycin.This paper could provide reference not only for the applier to improve the quality standard, but also for the peers to establish quality standard for compound chemical preparation which contains unstable principal components.
cephalexin; kanamycin monosulfate; purity of reagents; instability
韩宁宁,硕士,从事抗生素检验检测工作。E-mail: hnn19841001@126.com
赵晖。E-mail: 171977364@qq.com
2015-08-04
A
1002-1280 (2015) 11-0034-04
S859.79
