硝酸羟胺离子间相互作用的密度泛函理论研究
2015-03-08刘建国安振涛祁立雷满海涛王朝阳
刘建国, 张 倩,2, 安振涛,2, 祁立雷,2, 满海涛, 王朝阳
(1. 军械工程学院弹药工程系,河北石家庄050003; 2. 军械工程学院弹药保障与安全性评估军队
重点实验室,河北石家庄050003; 3. 华南师范大学化学与环境学院,广东广州510006)
硝酸羟胺离子间相互作用的密度泛函理论研究
刘建国1, 张倩1,2, 安振涛1,2, 祁立雷1,2, 满海涛1, 王朝阳3
(1. 军械工程学院弹药工程系,河北石家庄050003; 2. 军械工程学院弹药保障与安全性评估军队
重点实验室,河北石家庄050003; 3. 华南师范大学化学与环境学院,广东广州510006)
摘要:为研究硝酸羟胺离子间的相互作用,采用DFT-B3LYP/6-311++G(d, p)方法,对硝酸羟胺离子对的几何构型进行优化;通过自然集居分析计算了各原子的净电荷,并针对离子对体系形成过程中H原子的转移进行了理论研究;通过基组叠加误差(BSSE)和零点能校正(ZPEC)计算了离子对的相互作用能;用统计热力学方法对硝酸羟胺离子对在200~800K的热力学性质进行了计算。结果表明,离子对形成过程中H原子转移的反应势垒为46.605kJ/mol,离子对表现为HNO3和NH2OH之间相互作用的性质;构型II的相互作用能最大,为-48.145kJ/mol,稳定性排列顺序为构型II>构型III>构型IV;另外,构型II和构型III在常温下即可自发形成,构型IV只能在200K以下的低温才能自发形成。
关键词:量子化学;硝酸羟胺;离子对;相互作用;密度泛函理论;氢原子的转移;热力学性质
引言
硝酸羟胺是羟胺的硝酸盐,由还原组分羟胺和氧化组分硝酸组成[1-2]。硝酸羟胺基推进剂具有密度大、比冲高、安全、无毒等优点,广泛应用于炮弹发射、火箭推进、导弹姿态调控以及微型卫星的轨道调整[3-7]。目前,美国军方已研制出硝酸羟胺基发射药LP1846和LP1898,美国宇航局(NASA)路易斯研究中心在军方液体发射药的基础上开发出用于航天推进的HAN基单元推进剂[8]。
广义的分子间相互作用包括非极性分子、极性分子和离子间相互作用,分子间相互作用在高能体系的物理、化学和爆炸性质等研究中占有重要地位[9-10],对揭示其聚集状态、黏度、密度和物质间相互作用的本质等具有重要意义。居学海等[9]对硝仿肼离子对之间的相互作用进行了密度泛函理论研究,揭示了离子对之间相互作用的主要贡献为库伦作用;CAO Duan-lin等[11]采用密度泛函的方法对2,4-二硝基咪唑与甲醛的分子间相互作用进行了研究,并采用自然键轨道分析方法对其相互作用的本质进行了分析;WANG Zhao-xu等[12]对HCN和HNC之间的相互作用进行了研究,并对其热力学性质进行了计算。
本文采用DFT-B3LYP/6-311++G(d, p)方法,对硝酸羟胺离子对的几何构型、离子间的相互作用以及离子对的热力学性质进行研究,以期为硝酸羟胺基推进剂的制备及其性能的深入研究提供理论依据。
1计算方法
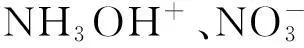
2结果与讨论
2.1硝酸羟胺离子对的几何构型优化

图以及硝酸羟胺离子对全优化几何构型和离子间距离Fig.1 Fully optimizad geometries and distance betweenNH3OH+,and ions of hydroxylamine nitrate ion pair
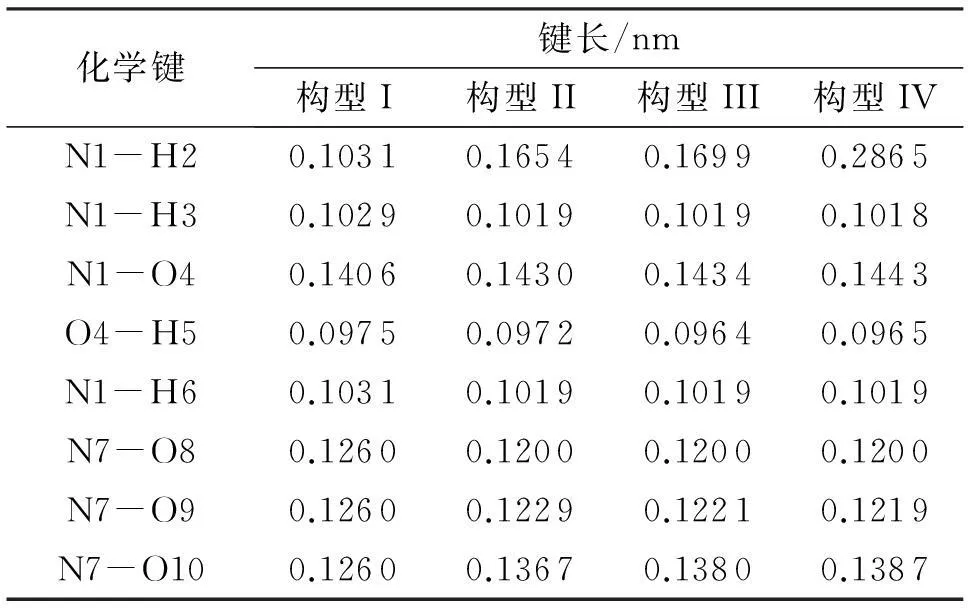
化学键键长/nm构型I构型II构型III构型IVN1-p0.10310.16540.16990.2865N1-p0.10290.10190.10190.1018N1-O40.14060.14300.14340.1443O4-H50.09750.09720.09640.0965N1-H60.10310.10190.10190.1019N7-O80.12600.12000.12000.1200N7-O90.12600.12290.12210.1219N7-O100.12600.13670.13800.1387
及硝酸羟胺离子对的键角
及硝酸羟胺离子对的二面角

随着键长的变化,硝酸羟胺的键角和二面角也发生了相应的改变。由图1和表2可以看出,与构型I相比,构型II的N1-O4-H5的键角减小了4.57°,O4-N1-H6的键角减小了6.86°;构型III的N1-O4-H5的键角减小了3.23°,O4-N1-H6的键角减小了7.18°;构型IV的N1-O4-H5的键角减小了2.6°,O4-N1-H6的键角减小了7.49°,其他键角在硝酸羟胺离子对的形成过程中变化较小。负离子上原子的二面角全部接近0或者180°,表明离子对形成过程中并没有对负离子的平面构型产生影响。但是,正离子的共面性在离子对形成过程中所受影响较大。如表3所示,构型II的p-N1-O4-H5的二面角增加了123.64°,H6-N1-O4-H5的二面角增加了61.047°;构型III的p-N1-O4-H5的二面角增加了123.96°,H6-N1-O4-H5的二面角增加了62.027°;构型IV的p-N1-O4-H5的二面角增加了127.39°,H6-N1-O4-H5的二面角增加了57.917°
2.2原子电荷和电荷转移
经自然集居分析所得各原子上的净电荷(Q)如表4所示。从表4可以看出,N原子和O原子带负电荷,呈负电性,H原子带正电荷,呈正电性。原子电荷变化较大的多是离子间距离较近的原子。与构型I相比,构型II的p净电荷增加了0.0830e,H5净电荷减少了0.0173e,与其形成离子间相互作用的N1净电荷增加0.3496e,O9净电荷减少了0.1329e;构型III的p净电荷增加了0.0899e,p净电荷减少了0.0941e,与其形成离子间相互作用的N1净电荷增加了0.3789e,O9净电荷减小了0.1752e;构型IV的p净电荷增加了0.0521e,H5净电荷减少了0.0096e,与其形成离子间相互作用的O4净电荷增加了0.3113e,O9原子净电荷减少了0.1716e。离子间相距较近的原子总电荷量呈增加的趋势,这是由于离子间相互吸引导致电荷向构型中心转移引起的。构型II、III、IV的偶极距分别为3.7890、0、4.1079、3.5928、4.2996 D。
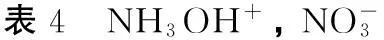
及硝酸羟胺离子对体系的
2.3离子间自然键轨道(NBO)相互作用
采用密度泛函B3LYP/6-311++G(d,p)方法对硝酸羟胺离子对体系进行自然键轨道(NBO)计算,得到了电子供体(Donor)轨道i、电子受体(Acceptor)轨道j及其稳定化能E,结果如表5所示。稳定化能E与相互作用强度之间成正比,稳定化能越大,i与j的相互作用强度越大[17]。从表5可以看出,构型II中N1的孤对电子第1对p-O10的σ反键轨道的稳定化能为194.43kJ/mol,构型III中的N1孤对电子第1对p-O10的σ反键轨道的稳定化能为160.21kJ/mol,构型IV中的O4孤对电子第2对p-O10的σ反键轨道的稳定化能为87.613kJ/mol。由此可以得出,构型II和III的离子间相互作用主要产生于阳离子的N的孤对电子与阴离子的H-O的反键轨道之间,构型IV的离子间相互作用主要产生于阳离子的O的孤对电子与阴离子的H-O反键轨道之间。另外,3个构型的稳定化能之和均在90kJ/mol以上,属于强相互作用,稳定化能排列顺序为构型II>构型III>构型IV。
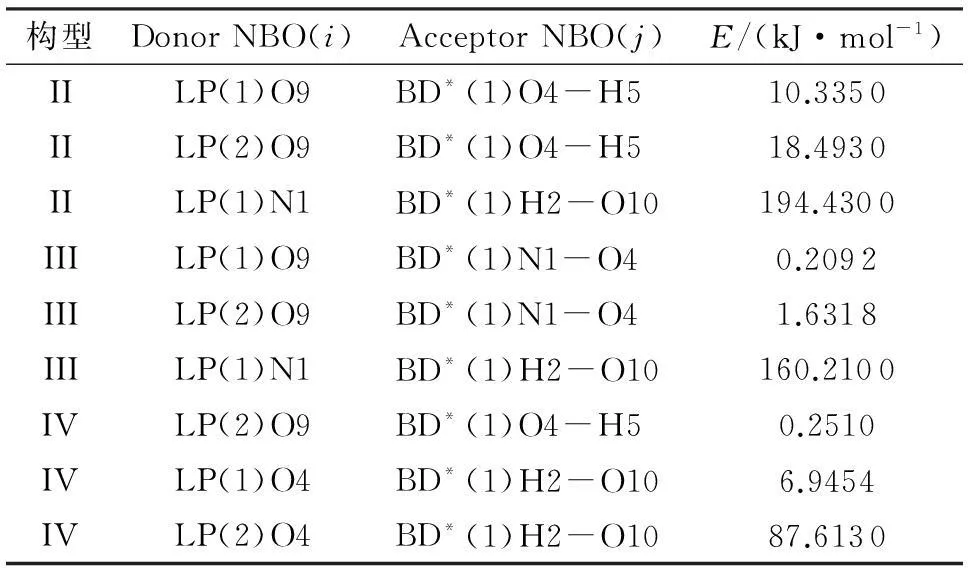
表5 B3LYP/6-311++G(d, p)水平上硝酸羟胺离子对
注:E为稳定化能;BD为成键轨道;BD*为反键轨道;LP为孤对电子;对于BD和BD*,(1)和(2)分别为σ轨道和π轨道;对于LP,(1)和(2)分别为第1和第2孤对电子。
2.4离子对形成过程中的氢转移
2.5相互作用能
在B3LYP/6-311++G(d, p)全优化构型下,将NH2OH、HNO3和各离子对体系的总能量以及经BSSE和ZPE校正前后的相互作用能的计算结果列于表6。从表6可以看出,构型II经校正后的结合能最大,为-48.145kJ/mol;构型IV经校正后的结合能最小,为-30.200kJ/mol。按照总能量、校正前以及校正后的相互作用能绝对值的排列顺序均为构型II>构型III>构型IV。由此可见,离子对体系稳定性的排列顺序为构型II>构型III>构型IV。另外,构型II、III和IV的ZPE校正值与BSSE校正值之和分别占经ZPE和BSSE校正后相互作用能的20.35%、20.00%和23.69%,说明进行ZPE和BSSE校正是必要的。

表6 B3LYP/6-311++G(d, p)水平上NH2OH、HNO3和硝酸羟胺离子对体系和的总能量、零点能和相互作用能
注:EHF为总能量;EZPE为零点能;ΔE为未校正的相互作用能;ΔE+BSSE为经基组叠加误差校正的相互作用能;ΔE+BSSE+ΔZPE为经基组叠加误差和零点能校正的相互作用能。
2.6热力学性质
同一温度下,焓变绝对值的顺序为:(ΔHT)II>(ΔHT)III>(ΔHT)IV。这进一步说明,构型II的相互作用强于构型III,构型III的相互作用强于构型IV。在不同温度下,随着温度的升高,ΔHT略有增加,说明温度升高离子间相互作用减弱。由ΔGT=ΔHT-TΔST可计算出各构型在不同温度下的ΔGT,从表7可以看出,298.15K时,构型II和III形成的过程中,ΔGT均为负值,表明在常温下该过程可以自发进行;然而,构型IV只能在200K以下的低温才能自发形成,这进一步说明了构型III的相互作用较弱。

表7 不同温度下NH2OH、HNO3以及硝酸羟胺离子对体系的热力学性质
3结论
(1)用Gaussian09软件在B3LYP/6-311++G(d,p)基组水平上对硝酸羟胺离子对相互作用进行了研究,硝酸羟胺离子对存在3种稳定的硝酸羟胺构型,构型II属于Cs点群,构型III和IV属于C1点群;经自然集居分析,离子间距离较近的原子电荷变化较大。这是由于离子间相互吸引导致电荷向构型中心转移,引起离子间相距较近的原子总电荷量增加。
(2)硝酸羟胺离子对形成过程中,首先发生H原子的转移,其转移势垒为46.605kJ/mol,3种构型不再表现为离子对之间的相互作用,而是表现为HNO3和NH2OH之间的相互作用。
(3)硝酸羟胺离子对体系相互作用能绝对值的排列顺序均为构型II>构型III>构型IV,由此得出,离子对体系稳定性的排列顺序为构型II>构型III>构型IV,其排列顺序与稳定化能的排列顺序相一致。
(4)对不同温度下的热力学性质研究表明,构型II和构型III在常温下即可自发形成,构型IV只能在200K以下的低温才能自发形成。
参考文献:
[1]Amrousse R, Katsumi T, Niboshi Y, et al. Performance and deactivation of Ir-based catalyst during hydroxylammonium nitrate catalytic decomposition[J]. Applied Catalysis A: General, 2013, 452: 64-68.
[2]LIU Li-jun, WEI Chun-yang, GUO Yu-yan, et al. Hydroxylamine nitrate self-catalytic kinetics study with adiabatic calorimetry[J]. Journal of Hazardous Materials, 2009, 162: 1217-1222.
[3]Barney G S, Duval P B. Model for predicting hydroxylamine nitrate stability in plutonium process solutions[J]. Journal of Loss Prevention in the Process Industries, 2011, 24: 76-84.
[4]Ulas A, Boysan E. Numerical analysis of regenerative cooling in liquid propellant rocket engines[J]. Aerospace Science and Technology, 2013, 24(1): 187-197.
[5]Fu Juan, Chen Xiao-qian, Huang Yi-yong. Validation of a compression mass gauge using groud tests for liquid propellant mass measurements[J]. Advances in Space Research, 2014, 53(9): 1359-1369.
[6]Dheeraj A, Basu P, Tharakan T J. Prediction of gas-core vortices during draining of liquid propellants from tanks[J]. Aerospace Science and Technology, 2014, 32(1): 60-65.
[7]Koh K S, Tengku F J C, Chik W K. Role of electrodes in ambient electrolytic decomposition of hydroxylammonium natrate (HAN) solutions[J]. Propulsion and Power Research, 2013, 2(3): 194-200.
[8]Amrousse R, Katsumi T, Itouyama N, et al. New HAN-based mixtures for reaction control system and low toxic spacecraft propulsion subsystem: thermal decomposition and possible thruster applications[J]. Combustion and Flame, 2015, 162(6): 2686-2692.
[9]居学海, 肖继军, 肖鹤鸣. 硝仿肼离子对相互作用的密度泛函理论研究[J]. 高等学校化学学报, 2003, 24(6): 1067-1071.
JU Xue-hai, XIAO Ji-jun, XIAO He-ming. DFT study of the intermolecular interaction of hydrazinium nitroformate ion pair[J]. Chemical Journal of Chinese Universities, 2003, 24(6): 1067-1071.
[10]候素青, 曹瑞林, 张文艳, 等. 氮杂杯[4]芳烃主体与RDX客体分子间相互作用的密度泛函理论[J]. 火炸药学报, 2008, 31(5): 19-23.
HOU Su-qing, CAO Duan-lin, ZHANG Wen-yan, et al. Density functional theory of intermolecular interactions of aza-calix[4] arene with RDX[J]. Chinese Journal of Explosives and Propellants, 2008, 31(5): 19-23.
[11]CAO Duan-lin, REN Fu-de, WANG Jian-long, et al. Theoretical study on intermolecular interactions of 2,4-dinitroimidazole with methanol[J]. Journal of Molecular Structure: Theochem, 2007, 805: 53-60.
[12]WANG Zhao-xu, ZHANG Jing-chang, WU Jun-yong, et al. Theoretical investigation on intermolecular interactions between HCN and HNC: the nature and thermodynamic properties[J]. Journal of Molecular Structure: Theochem, 2007, 806: 239-246.
[13]Alavi S, Thompson D L. Effects of alkyl-group substitution on the proton-transfer barriers in ammonium and hydroxylammonium nitrate salts[J]. J Phys Chem A, 2004, 108: 8801-8809.
[14]Arjunan V, Kalaivani M, Marchewka M K, et al. Crystal structure, vibrational and DFT simulation studies of melaminium dihydrogen phosphite monohydrate[J]. Journal of Molecular Struture, 2013, 1045: 160-170.
[15]LI Lai-cai, HU Feng, CAI Wan-fei, et al. Density functional theory study on hydrogen bonding interaction of luteolin-(H2O)n[J]. Journal of Molecular Structure: Theochem, 2009, 911: 98-104.
[16]何春芳, 叶近婷, 高阳, 等. 三聚磷酸钠与柠檬酸钠钙螯合机理和螯合能力的对比分析[J]. 分子科学学报, 2015, 31(3): 198-202.
HE Chun-fang, YE Jin-ting, GAO Yang, et al. Comparative analysis of calcium chelation mechanism and chelating ability about sodium tripolyphosphate and citric acid sodium[J]. Journal of Molecular Science, 2015, 31(3): 198-202.
[17]YUAN Mei-rong, LI Zhou-min. Crystal structure and DFT studies of N1, N6-Di (9H-fluoren-9-ylidene) hexane-1,6-diamine[J]. Journal of Molecular Structure, 2013, 1031: 263-268.
Density Functional Theory Study on Intermolecular Interactions of
Hydroxylamine Nitrate Ion Pair
LIU Jian-guo1, ZHANG Qian1,2, AN Zhen-tao1,2, QI Li-lei1,2, MAN Hai-tao1, WANG Chao-yang3
(1. Department of Ammunition Engineering, Ordnance Engineering College, Shijiazhuang 050003, China;
2. Military Key Laboratory for Ammunition Support and Safety Evaluation, Ordnance Engineering College,
Shijiazhuang 050003, China; 3. School of Chemistry and Environment, South China Normal
University, Guangzhou 510006, China)
Abstract:To study the interactions of hydroxylamine nitrate ion, the geometry of hydroxylamine nitrate ion pair was optimized using the B3LYP/6-311++G (d, p) method. The atomic net charge was calculated by natural population analysis and the theoretical research of H atom transfer in the process of ion pair system formation was carried out. The interaction energy of ion pair was calculated by basis set superposition error (BSSE) and zero point energy correction (ZPEC). The thermodynamic properties of hydroxylamine nitrate ion pair in the temperature range of 200 to 800K were calculated using the statistical thermodynamic method. The results show that the reaction barrier of H atom transfer in the process of ion pair formation is 46.605kJ/mol. The ion pair manifests as the nature of interactions between HNO3and NH2OH. The interaction energy of the configuration II is greatest, as -48.145kJ/mol, and the stability decreases in the order of configuration II>configuration III>configuration IV. Configurations II and III can be spontaneously formed at the room temperature, while configuration IV can only be formed spontaneously at lower temperature below 200K.
Keywords:quantum chemistry; hydroxylamine nitrate; ion pair; interaction; density functional theory; H atom transfer; thermodynamic property
通讯作者:张倩(1974-),女,副教授,从事含能材料的合成与分子模拟等研究。
作者简介:刘建国(1988-),男,博士研究生,从事含能材料的合成与分子模拟等研究。
基金项目:“十二五”装备预研项目(40404010303)
收稿日期:2015-09-09;修回日期:2015-10-14
中图分类号:TJ55; O641.121
文献标志码:A
文章编号:1007-7812(2015)06-0039-06
DOI:10.14077/j.issn.1007-7812.2015.06.008
