一个遗传性非综合征型耳聋家系的GJB2基因突变分析*
2015-03-04王义旺胡祥上全庆丽姜海鸥
王义旺,胡祥上,全庆丽,姜海鸥△
(1.湖南医药学院附属医院/怀化市第一人民医院耳鼻喉科,湖南怀化418000;2.湖南医药学院医学遗传学教研室,湖南怀化418000)
耳聋是最常见的感觉障碍性疾病之一,在新生儿中发病率约为1/1 000。按表型特征不同,遗传性耳聋可分为综合征型耳聋(syndromic hearing loss,SHL)和非综合征型耳聋(nonsyndromic hearing loss,NSHL)[1]。NSHL可表现为多种遗传方式,包括常染色体显性遗传,常染色体隐性遗传,X 连锁遗传,Y 连锁遗传和线粒体遗传等。遗传性耳聋具有高度的遗传异质性。迄今为止,共定位NSHL 基因位点136个,已克隆的致病基因有60余个,其中导致遗传性NSHL的基因中,GJB2、SLC26A4、MYO15A、OTOF、CDH23和TMC1等基因突变较为常见,其中又以GJB2 基因突变最为常见[2],在我国约18.31%的耳聋患者是由GJB2 基因突变导致的[3]。鉴此,作者对1个遗传性NSHL 家系成员的GJB2基因编码序列进行分析,旨在寻找耳聋患者的致病基因突变。
1 资料与方法
1.1 一般资料 该家系4代共14人,其中耳聋患者4例(图1)。先证者(Ⅲ2),男,42岁,12岁起双侧耳鸣并进行性听力减退,36岁发展为全聋。纯音测听显示左耳听力为85分贝,右耳听力为80分贝,属于重度耳聋。除耳聋之外,不伴其他器官系统的异常。该家系耳聋患者的起病年龄为12~25 岁,其他患者与先证者临床症状相似。临床表现为双侧耳鸣,听力减退为双耳对称性、以高频听力下降为主的感觉神经性耳聋。耳部CT 扫描、前庭功能及全身体格检查,均未发现异常。
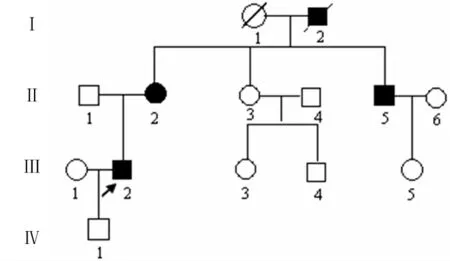
图1 患者家系图

表1 PCR 扩增GJB2基因的引物
1.2 方法
1.2.1 DNA 制备 在获得知情同意之后,采集该家系9个成员的外周血样本,包括3例患者(Ⅱ2、Ⅱ5、Ⅲ2)和6名健康人(Ⅱ1、Ⅱ3、Ⅱ6、Ⅲ1、Ⅲ5、Ⅳ1)。血液基因组DNA 提取试剂盒购自北京天根生化公司(批号DP318),按产品说明书介绍的方法提取基因组DNA。DNA 浓度测定后稀释成为25ng/μL 工作液。
1.2.2 PCR 扩增应用Primer5软件设计并合成3对引物(表1),扩增区域覆盖GJB2基因的两个外显子及外显子与内含子交界区域。PCR 反应体系:总体积为15μL,2×GC buffer 7.50μL(含MgCl210 mmol/L),dNTP 混合液(10 mmol/L)0.75μL,正反向引物(10μmol/L)各0.75μL,模板基因 组DNA 0.03μg,rTaq DNA 聚合酶(5U/μL)0.80μL,双蒸水补至15.00μL。反应条件:95 ℃预变性10min、95 ℃变性30s、55~60℃退火30s、72℃延伸45s,35个循环,72℃再延伸10 min。PCR 产物经1%琼脂糖凝胶,恒压140V 电泳25min,溴化乙锭/紫外线观察,以证实PCR 扩增的片段。
1.2.3 产物纯化、序列测定及分析 PCR 产物纯化(生工SK1141)后送至上海生工生物工程技术服务有限公司,在ABI3730自动测序仪上应用相应的PCR 引物进行双向直接序列测定,测序结果使用DNAStar软件进行分析。发现的序列变异在NCBI和Hapmap的多态数据库中进行查询,确定是否为单核苷酸多态(single nucleotide polymorphism,SNP)。通过同时检测家系内所有患者及其健康同胞的相应序列,鉴定变异序列是否与疾病表型共分离。
2 结 果
该家系共有存活耳聋患者3例,从家系图分析,符合常染色体显性遗传;所有患者均为语后聋,为双耳对称性、进行性、高频听力下降为主的感觉神经性聋,随年龄增加耳聋程度逐步加重,最终进展为全频受累的重度感音神经性聋。患者出现听力下降的年龄,从12~25岁。Ⅱ2,女,65岁,16岁出现听力下降,48岁发展为全聋,目前左耳听力为75分贝,右耳听力为80分贝。Ⅱ5,男,55岁,25 岁出现听力下降,42 岁发展为全聋,目前左耳听力为85分贝,右耳听力为84分贝。
直接双向测序后,发现GJB2基因的6个变异序列(表2、图2),这些变异既存在于部分患者中,也在健康亲属中检测到,与疾病不存在共分离。本研究中发现的GJB2基因的核苷酸改变是SNP。

图2 GJB2基因中检测到的核苷酸变异序列

表2 GJB2基因中检测到的变异序列
3 讨 论
耳聋是导致交流障碍最常见的疾病,大多数发病都与遗传有关。遗传性耳聋患者中70%以上为NSHL。耳聋具有高度的遗传异质性,相关基因众多,但在不同人群中GJB2基因突变均是导致NSHL的最主要致病基因[4]。Mignon 等[5]于1996年将人类GJB2定位于13q11-q12,基因全长5 510bp,含两个外显子,而编码序列位于exon2,长为681bp。GJB2含有226个氨基酸的蛋白质,相对分子质量约为26×103,也称缝隙连接蛋白26(connexin 26,Cx26)。Cx26是一种跨膜蛋白,4次跨膜,共分5个区:N 端区、跨膜区、细胞外区、胞浆内连接区和C端区。Cx26蛋白以六聚体形式结合形成缝隙连接,形成耳蜗内钾离子运输通道,以维持耳蜗内较高水平电位[6]。GJB2基因突变可导致Cx26 蛋白功能缺陷,减少钾离子再循环效率,进而导致听力损害[7]。目前发现,GJB2至少有111种突变可以引起NSHI,其中显性突变9 种,隐性突变92 种,遗传模式不明突变10种。
本研究的NSHL家系中,所有患者均为语后聋,为双耳对称性、进行性、高频听力下降为主的感觉神经性聋,不伴其他器官系统的异常。根据其临床特征和遗传方式,作者认为该家系属于常染色体显性遗传的非综合征型耳聋。进一步用分子遗传学方法尝试了GJB2 基因的突变筛查,寻找患者特有的突变。检测到的6个变异序列既存在于患者中,也出现在患者的健康亲属中,未发现与疾病表型共分离的变异。其中c.79G>A(p.Val27Ile)和c.341G>A(p.Glu114Gly)为已报道的核苷酸多态,而位于3′-UTR 的g.4159T>C、g.5142G/T、g.5227G/A、g.5352T/C突变为本研究国内外首次报道。尽管本研究在GJB2基因的编码区发现了2个杂合突变79G>A(p.Val27Ile)和341G>A(p.Glu114Gly),但在该家系患者和健康人中均存在这些突变,因此,作者认为它们更可能是该基因的SNP,这与大多数研究者认为杂合突变79G>A 和341G>A是GJB2基因的多态相一致的[8-10]。所以,作者初步排除了GJB2为本研究NSHL家系的致病基因,说明遗传性非综合征型耳聋存在明显的遗传异质性。
[1] Walsh T,Pierce SB,Lenz DR,et al.Genomic duplication and overexpression of TJP2/ZO-2leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51[J].Am J Hum Genet,2010,87(2):101-109.
[2] Walsh T,Shahin H,Elkan-Miller T,et al.Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2as the cause of nonsyndromic hearing loss DFNB82 [J].Am J Hum Genet,2010,87(1):90-94.
[3] Xiao ZA,Xie DH.GJB2(Cx26)gene mutations in Chinese patients with congenital sensorineural deafness and a report of one novel mutation[J].Chin Med J(Engl),2004,117(20):1797-1801.
[4] Kim HJ,Park CH,Kim HJ,et al.Sequence variations and haplotypes of the GJB2gene revealed by resequencing of 192chromosomes from the general population in Korea[J].Clin Exp Otorhinolaryngol,2010,3(1):65-69.
[5] Mignon C,Fromaget C,Mattei MG,et al.Assignment of connexin 26(GJB2)and 46(GJA3)genes to human chromosome 13q11-q12and mouse chromosome 14D1-E1by in situ hybridization[J].Cytogenet Cell Genet,1996,72(2/3):185-186.
[6] Su CC,Li SY,Su MC,et al.Mutation R184Qof connexin 26in hearing loss patients has a dominant-negative effect on connexin 26and connexin 30[J].Eur J Hum Genet,2010,18(12):1061-1064.
[7] Terrinoni A,Codispoti A,Serra V,et al.Connexin 26(GJB2)mutations as a cause of the KID syndrome with hearing loss[J].Biochem Biophys Res Commun,2010,395(1):25-30.
[8] Kelley PM,Harris DJ,Comer BC,et al.Novel mutations in the connexin 26gene(GJB2)that cause autosomal recessive(DFNBl)hearing loss[J].Am J Hum Genet,1998,62(4):792-799.
[9] Abe S,Usami S,Shinkawa S,et al.Prevalent counexin 26 gene(GJB2)mutations in Japanese [J].J Med Genet,2000,37(1):41-43.
[10] 肖自安,冯永,潘乾.非综合征性耳聋患者连接蛋白26基因突变的研究[J].中华耳鼻咽喉科学杂志,2000,35(3):188-191.
