作为2型糖尿病治疗药物的G蛋白偶联受体40激动剂研究进展
2015-02-01周牧星杨凌云周金培张惠斌
周牧星,杨凌云,周金培,张惠斌
(中国药科大学江苏省抗代谢性疾病药物重点实验室,江苏 南京 210009)
作为2型糖尿病治疗药物的G蛋白偶联受体40激动剂研究进展
周牧星,杨凌云,周金培,张惠斌*
(中国药科大学江苏省抗代谢性疾病药物重点实验室,江苏 南京 210009)
2型糖尿病约占糖尿病总病例数的90%,目前研发的其新型治疗药物主要是通过调节糖代谢通路来控制血糖水平,它们可通过激活G蛋白偶联受体尤其是G蛋白偶联受体40,增强胰岛β细胞功能,促进胰岛素分泌,提高机体对胰岛素的敏感性,从而达到治疗糖尿病的目的。G蛋白偶联受体40作为抗2型糖尿病的新靶点,以其潜在优势,在糖尿病治疗领域备受关注。简介G蛋白偶联受体与其配体游离脂肪酸,重点综述不同结构的G蛋白偶联受体40激动剂的研究进展。
G蛋白偶联受体40;激动剂;2型糖尿病; 胰岛素分泌; 血糖水平
糖尿病是一种严重威胁人类健康的疾病,其中2型糖尿病约占糖尿病总病例数的90%。2型糖尿病是非胰岛素依赖的糖尿病,其发病机制主要是肝糖原分泌过剩、胰岛素抵抗和胰岛β 细胞的功能性障碍[1]。最近几年,针对2型糖尿病的治疗研究主要集中在通过调节糖代谢通路来促进胰岛素的分泌,控制血糖水平,如利用胰高血糖素样肽-1(GLP-1)类似物以及二肽基肽酶-4(DPP-4)抑制剂等的治疗方案。此外,还有一些化合物能够通过与G 蛋白偶联受体(GPR)结合,促进胰岛素的分泌,从而达到治疗2 型糖尿病的目的,其中包括GPR40 激动剂等。本文重点综述GPR40 激动剂的研究进展。
1 G蛋白偶联受体与游离脂肪酸
GPR为一类细胞表面受体,其对光线、气味、神经信号和荷尔蒙等多种胞外信号敏感,因而可产生一系列胞内信号。据估计,人类GPR大约有850种,它们在特定的细胞或组织中表达,参与各种生理作用[2]。约有30%药物的靶点是GPR,并且可能有更多的GPR成为潜在的药物靶点,因此对GPR的鉴定和分类尤为重要[3]。人类和鼠类的GPR具有高度同源性,这表明这些受体在生物进化过程中并未受到影响,可发挥一定的生理作用。由于因人类GPR突变而引起的疾病很少,因此GPR可作为一个稳定的治疗靶点[4]。
在过去的10年中,对GPR分子药理学方面的研究取得了重要进展,研究发现GPR119、GPR84、GPR120、GPR40 (FFAR1)、GPR43 (FFAR2) 和 GPR41 (FFAR3)等受体的配体是各种游离脂肪酸(FFA),这些对FFA敏感的GPR已成为新药研发的靶点[5]。
FFA可按脂肪链的长度分类:含6个以下碳原子的称短链FFA,含6~12个碳原子的称中链FFA,含12个以上碳原子的称长链FFA。FFA可作为分子受体和基因表达的传导介质,其中,中、长链FFA可激动GPR40和GPR120,长链FFA还可激动GPR119,中长链FFA则能激动GPR84,而短链FFA可激动GPR43和GPR 41[6]。
FFA是人体必须的营养素,同时也在各种生理作用的信号传导中充当信号分子,而过氧化物酶体增殖物激活受体 (PPAR)和脂肪酸结合蛋白(FABP)被认为是FFA的受体。然而,在最近的10年中,对GPR的进一步研究发现了一系列FFA受体。GPR是一种七次跨膜受体,能通过异源三聚体配体间的作用激动G蛋白,从而在各种生理作用中扮演重要角色。在FFA受体中,FFAR2和FFAR3是短链FFA受体,而FFAR2主要在免疫细胞中表达,它在内环境稳态和炎症反应中发挥多种作用[7],FFAR3则主要在脂肪组织和胃肠道中表达。短链FFA激动FFAR3,可引起瘦素分泌,表明FFAR3可能在能量稳态中发挥作用。
在肥胖症和2型糖尿病患者中,血浆FFA浓度升高,导致脂肪堆积和靶组织中胰岛素抵抗。FFA在胰岛β细胞中发挥多种作用,其急性作用可促进胰岛素分泌,而长期作用可能会损害胰岛素分泌,这种双重作用表明FFA在2型糖尿病引发高血糖和低胰岛素的过程中均发挥作用。因此,在2型糖尿病治疗领域,对FFA及其受体GPR的研究成为一大热点[8-9]。
2 G 蛋白偶联受体40激动剂
人类GPR40基因在人类基因库U62631片段,与GPR41、GPR42和GPR43一同位于染色体19q13.1上,出现在CD22基因区域的下游,这个区域与2型糖尿病家族的高三酰甘油血症有关。GPR40与GPR41和GPR42的同源性较高,与GPR43的同源性不高。GPR40是有七次跨膜结构特征的鸟苷三磷酸(GTP)结合蛋白偶联受体[10],主要表达于胰岛β细胞,也在胃肠道的分泌细胞中表达,可激活GLP-1受体和促进葡萄糖依赖性的促胰岛素多肽分泌等。FFA对胰岛素分泌的放大作用如下:葡萄糖浓度的升高加速细胞内葡萄糖的代谢,导致胞液中腺苷三磷酸/腺苷二磷酸(ATP/ ADP)水平上升,胞膜上ATP依赖的钾离子通道(KATP)关闭,胞膜去极化,L型Ca2+通道打开;随后,FFA刺激胞膜上七次跨膜受体GPR40,通过磷脂酰肌醇信号转导途径,刺激内质网释放Ca2+,并进一步打开L型Ca2+通道,引起细胞外Ca2+内流,胞内Ca2+浓度大大升高,从而促使胰岛素分泌[11](又见:Sawzdargo等, Biochem Biophys Res Commun, 1997年)。
GPR40激动剂的结构类型较多,其中主要有两类:对氨基苯丙酸衍生物和对羟基苯丙酸衍生物。
2.1 对氨基苯丙酸衍生物
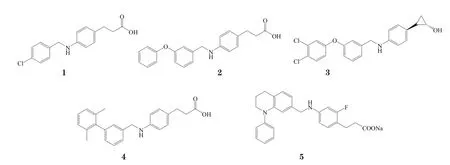
有研究者以2-(4-(烷基)氨基苯)丙酸为先导化合物,通过高通量筛选,发现化合物1为一小分子GPR40激动剂,其结构与月桂酸相似,对GPR40的pEC50为6.03,表现出较好的水溶性(0.83 g·L-1),具较低的相对分子质量(Mr=289),logP为3.6;而且,化合物1经结构改造,其活性可提高100倍,但其羧酸部分被酰胺取代后,活性会降低。进一步的构效关系研究表明化合物1的活性和其羧酸部分的性质有关,且通过对其苯丙酸的两个碳原子进行改造,发现了更多的活性类似物[12-13]。
GW9508(2)对GPR40具有较好的激动活性(pEC50=7.19),且对GPR120也有一定的激动作用(pEC50=5.46),不过其对GPR40的选择性是对GPR120的100倍,但对GPR41和GPR43无活性。且研究表明,GW9508的体内外活性均较高,生物利用度较好(65%),半衰期较长(口服半衰期为5.9 h,静注半衰期为5.3 h);此外, GW9508有适度的水溶性(0.29 g·L-1),对细胞色素450(CYP450)无抑制作用[13]。
葛兰素史克公司研究人员发现,对GW9508中芳香环和苯丙酸部分进行改造,还可获得其他一系列对氨基苯丙酸衍生物,其中具代表性的活性化合物3对GPR40的pEC50为8.5。武田公司也报道了一个对氨基苯丙酸衍生物(4),其拥有联苯结构,而不是苯氧基结构,它对GPR40的pEC50为7[13]。阿斯特拉公司研究人员则发现了一个新的对氨基苯丙酸衍生物(5),其对GPR40的pEC50为7.72;且构效关系研究表明,对氨基苯丙酸和四氢喹啉相连,有利于该类化合物对GPR40的激动作用[14]。
2.2 对羟基苯丙酸衍生物
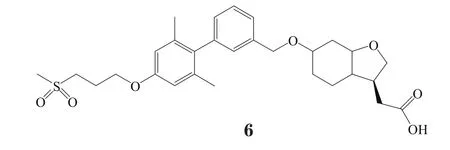
对羟基苯丙酸类GPR40激动剂中进入Ⅲ期临床研究的有武田(Takeda)公司开发的TAK-875(6),其半衰期为28~30 h[15-16]。最新的为期24周的Ⅲ期临床试验数据表明,2型糖尿病患者接受TAK-875(25或50 mg,qd)治疗后,第2周即出现空腹血糖降低。且TAK-875和格列美脲或西他列汀的临床疗效比较以及与西他列汀联用的疗效也被研究考察过[17-19]。然而,由于对肝毒性的担忧,武田公司于2013年12月终止了对TAK-875的开发。
由TAK-875与GPR40受体结合的复合物晶体X衍射图(见图1)可见,TAK-875与GPR40的跨膜螺旋3~5之间非经典部分以及胞外环2(ECL2)结合,这个结合部位靠近细胞膜外表面;TAK-875是一个变构结合调节剂,结合位点是一个变构结合口袋;TAK-875的羧基部分与GPR40上多个精氨酸和酪氨酸残基相互作用。不过,研究发现,GPR40上单个精氨酸残基发生突变,并未导致TAK-875的活性显著下降,表明与GPR40结合的某些精氨酸残基对受体活性无影响。但这些氨基酸残基组成的电性口袋保持了整个受体处于未激活状态,一旦电性口袋被干扰,受体即被激活。TAK-875侧链部分暴露在受体之外,对化合物与受体的结合影响不大,结构容忍性较强,修饰改造的空间很大;而砜基的存在致使TAK-875的亲脂性降低,改善了化合物在体内的吸收、分配、代谢、排泄和毒性(ADMET)性质[20]。
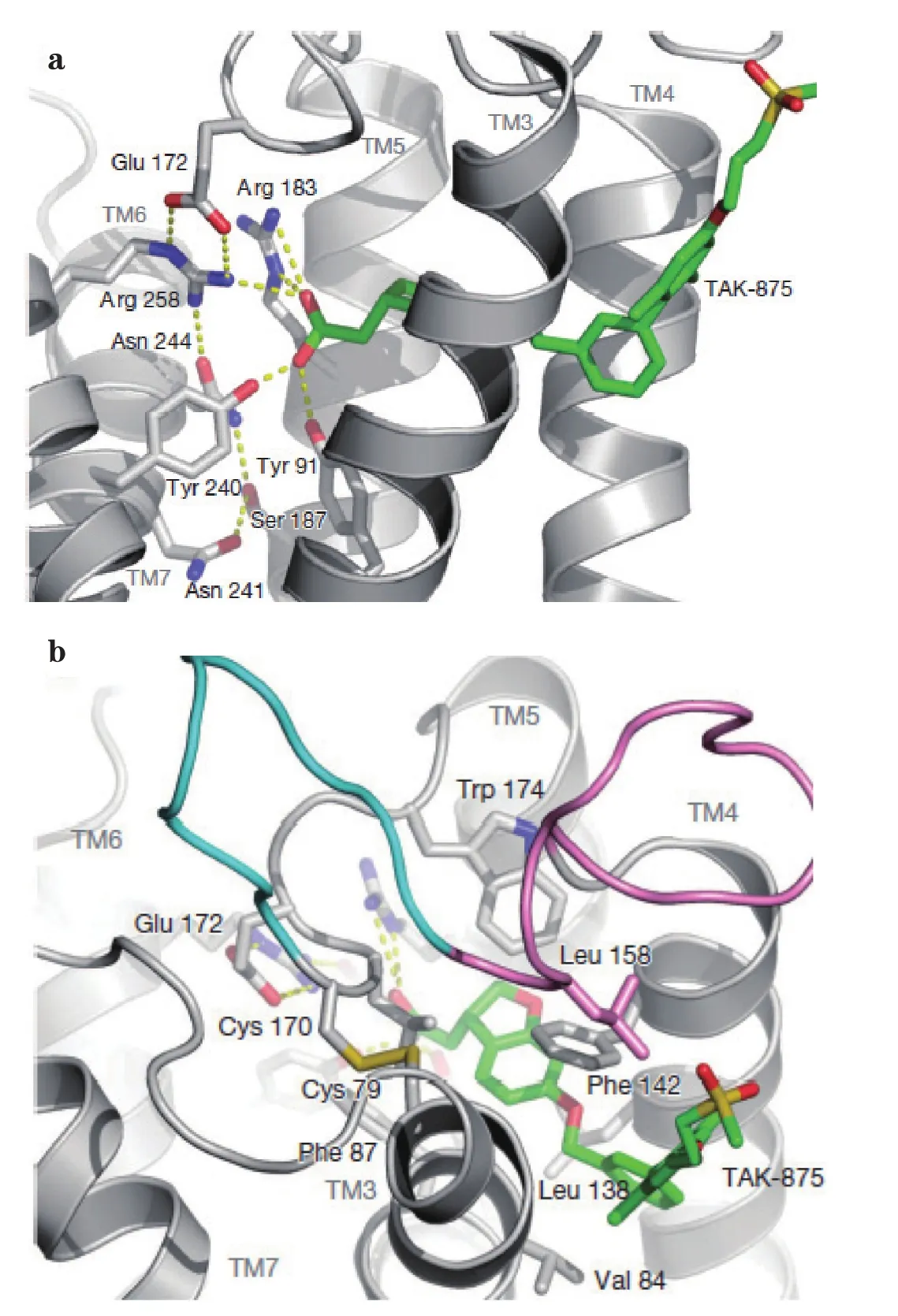
图1 TAK-875与GPR40的复合物晶体X衍射图Figure 1 Crystal X-ray diffraction diagram of TAK-875-GPR40 complex
依据图2和一系列相关化合物的研究报道,笔者对此类化合物的构效关系进行了如下推断:1)C环为苯环较为适宜,替换为芳杂环后,会导致化合物活性降低;2)X为连接臂,2个原子长度时,可使化合物活性达到最佳;3)R1为卤素及较小的烷基取代时,化合物活性最佳;4)C环和R2呈苯丙酸结构时,化合物活性最佳;5)R2基团α位引入取代基或环状结构时,可避免化合物被氧化,致其半衰期延长;6)R3为亲水性的侧链或环状结构时,可改善化合物的药代动力学性质,使其活性提高。
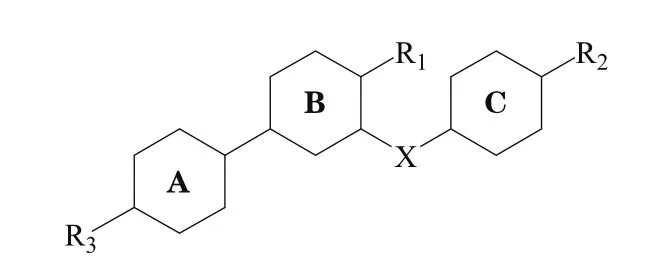
图2 对羟基苯丙酸类化合物构效关系示例Figure 2 Example of structure-activity relationship for p-hydroxy benzoic acid compounds
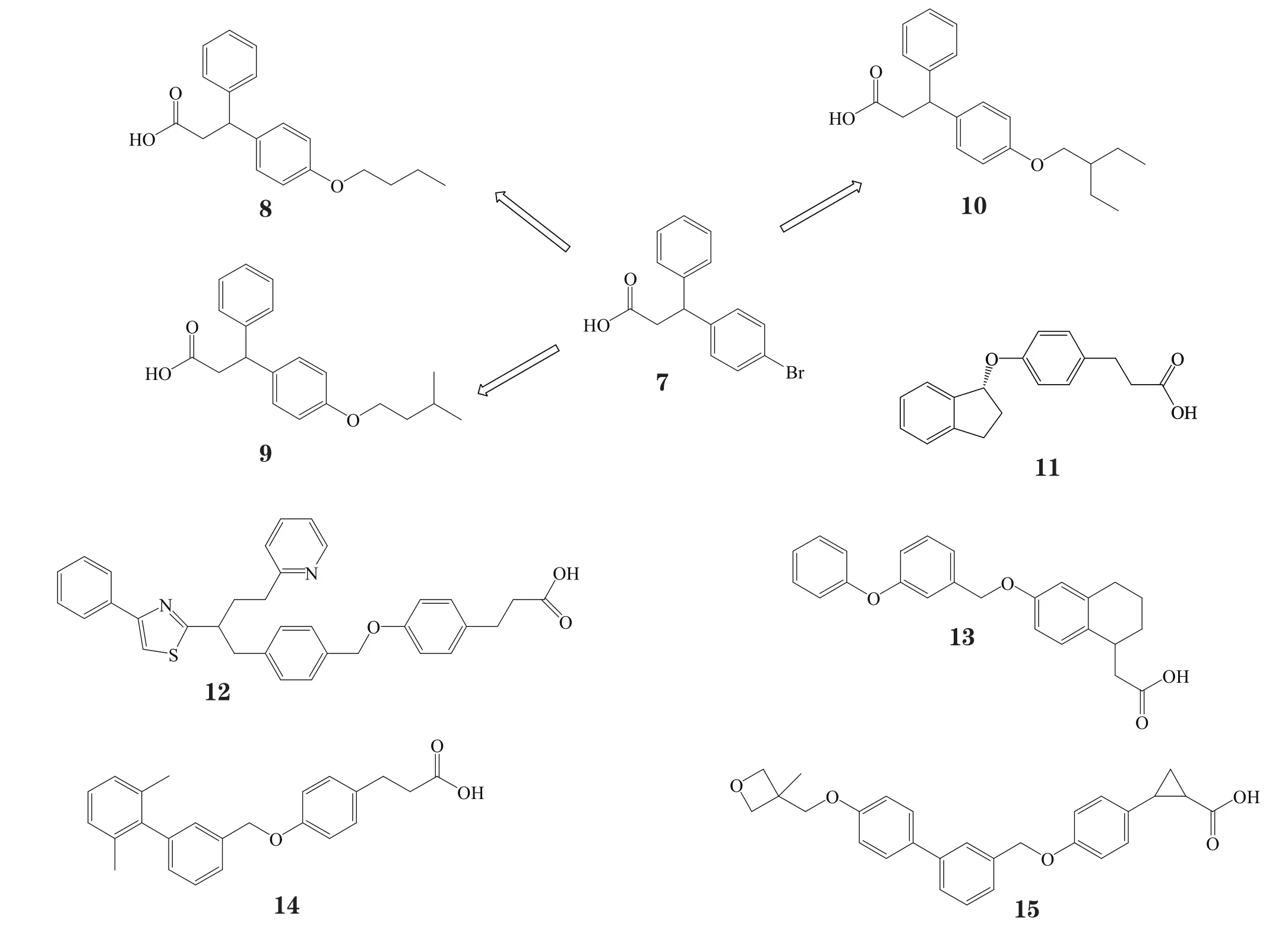
在此类化合物的研究初期,对化合物的修饰主要是在R2保持苯丙酸结构的同时,对A和B环进行结构改造,从而获得一系列3-(4-烷氧苯基)苯丙酸结构的衍生物。且研究发现,这类衍生物结构中苯丙酸的3位适宜与另一取代苯直接相连,若其间相隔1~3个碳原子,化合物则将失去活性。由此,经高通量筛选得到的化合物7为GPR40的温和激动剂(pEC50为 5.04),对其进一步修饰改造,即获得一类具有毫摩尔级活性的新型化合物,如化合物8、9、10。其中,活性最好的为化合物8,实验显示,它具有良好的生物利用度(87%)和半衰期(6.33 h),且口服吸收迅速(0.5 h),血药浓度可达4 758 μg · L-1,值得进一步研究开发[21]。
有研究者将双环或联苯结构等与对羟基苯丙酸骨架相连得到一系列新的化合物,其中,化合物11~14对GPR40的pEC50大于8,但化合物12在结构上与其他化合物不同,其碳链长度增加,活性有所降低,而10 μmol·L-1的化合物14对GPR40的激动活性为相同浓度FFA的107%~181%,化合物15则在相同浓度下的激动活性为内源性激动剂的119%[22]。
体内实验发现,对羟基苯丙酸衍生物因苯丙酸β位易发生β氧化而失去活性,半衰期较短。因此,有研究者尝试对此类化合物中苯丙酸β位进行取代修饰或在β位引入环状结构,以改善其半衰期。
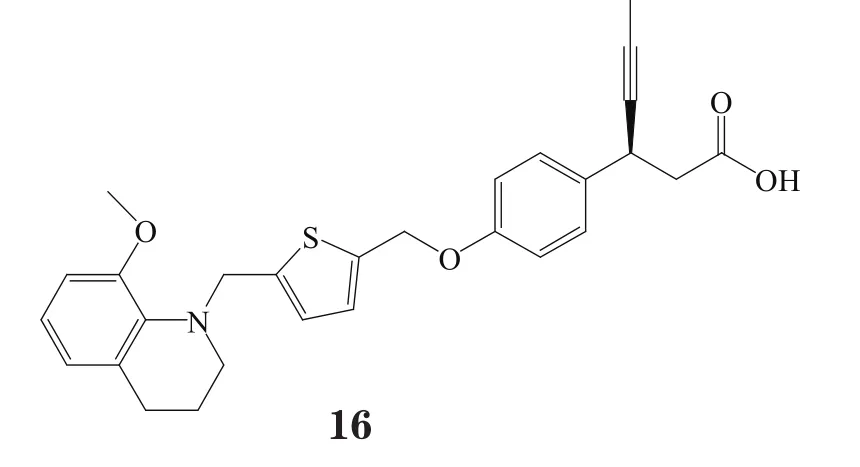
LY2881835(16)即是由此通过高通量筛选而发现的一个选择性强效GPR40激动剂(EC50=233 nmol·L-1),其活性为亚麻油酸的91%;且其在人和大鼠体内以及小鼠胰岛瘤MIN6细胞中均能强效促进新胰岛素分泌,在腹腔注射的葡萄糖耐受实验(ipGTT)中,其对正常小鼠可产生剂量依赖性降糖作用(ED90= 0.58 mg·kg-1)[23-24]。在2011年,礼来公司对LY2881835进行了Ⅰ期临床试验,考察了其临床副作用[25]。可是,之后未见有关此化合物的进一步报道。
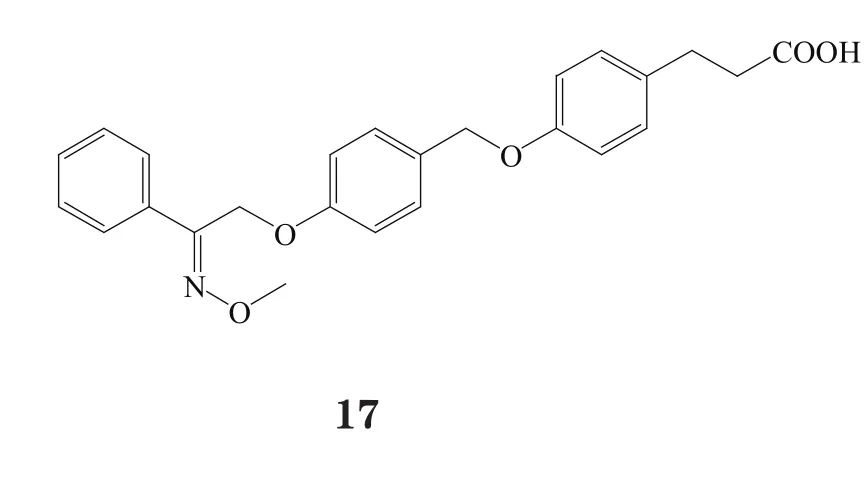
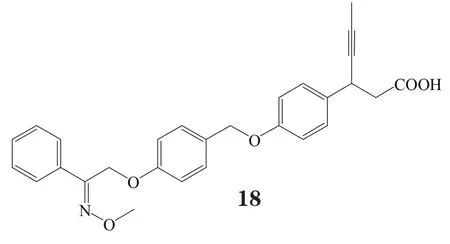
2013年,Connexios公司使用选择性GPR40激动剂CNX-011-67(EC50= 0.24 nmol·L-1)对ZDF大鼠进行7周长期性实验研究,结果表明,经口给予该化合物,可极大促进胰岛素分泌,延迟空腹高血糖,改善外周组织中胰岛素通路,降低血清FFA和三酰甘油水平。但此化合物的结构尚未公开。至今该公司针对GPR40激动剂类化合物的专利只有一个,此专利化合物的特点是具有N-甲基亚胺取代结构,该结构使用苯丙酸或其生物电子等排体作为酸性头部,如化合物17~20[26],其中大部分化合物激动GPR40的EC50小于10 nmol·L-1,此类化合物的活性与其苯丙酸β位的手性相关[15,27-30]。不过,该公司也未公开此类化合物是GPR40的完全激动剂或部分激动剂。
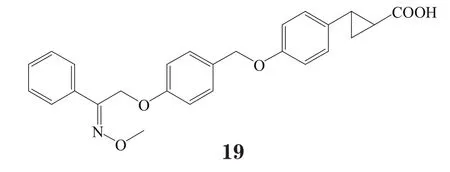
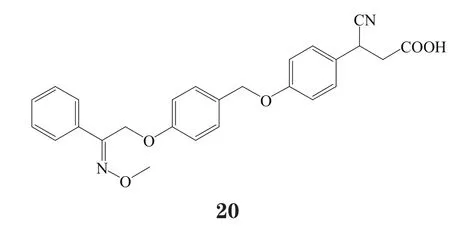
有研究者在对对羟基苯丙酸衍生物进行一系列的系统结构优化后发现了化合物21和22,这两个化合物在体外实验中均表现出对GPR40的良好激动活性与药理作用(EC50分别为71和130 nmol·L-1),其中化合物21在小鼠口服糖耐量实验中,以10 mg·kg-1剂量给药,极大地促进了胰岛素分泌。然而,化合物22易脱靶而产生对CYP2C9、hERG以及CaV1.2通道的抑制作用[16]。
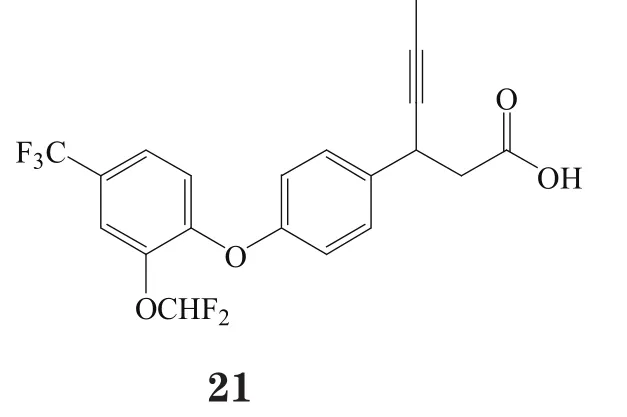
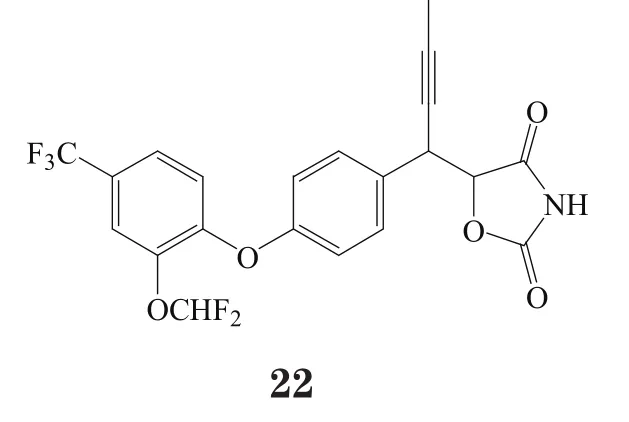
AMG-837(23)是第一个进入Ⅰ期临床试验的GPR40激动剂,一系列的临床前体内和体外研究证明,其是一个GPR40部分激动剂,具有良好的药理作用。Ⅰ期临床试验显示,AMG-837对健康受试者无降糖作用,证明它无低血糖副作用[31]。然而,由于其他安全性问题,AMG-837没能被进一步开发。
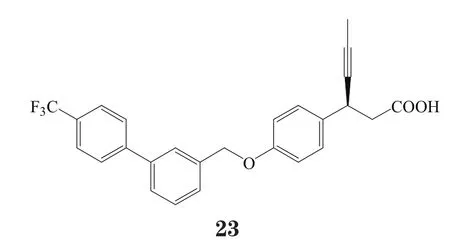
JTT-851是日本烟草公司开发的GPR40激动剂类化合物,目前处于Ⅱ期临床开发阶段,其结构尚未公开[32-33]。不过,该公司报道了另一GPR40激动剂化合物(24),其对GPR40的EC50为10~100 nmol·L-1[34]。
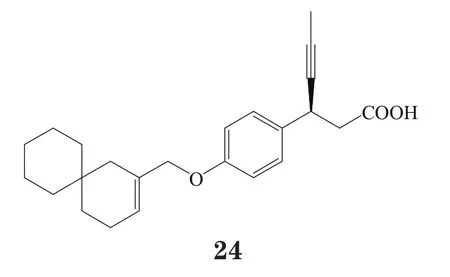
另一个进入Ⅰ期临床试验的此类化合物是印度Piramal公司开发的p11187,虽然其结构目前还未公开,但根据最近公开的该公司申请的此化合物专利,推测其可能是该公司将武田公司开发的化合物改造而来的化合物25,其对人GPR40具有良好的激动活性(EC50<100 nmol·L-1)[35-36]。
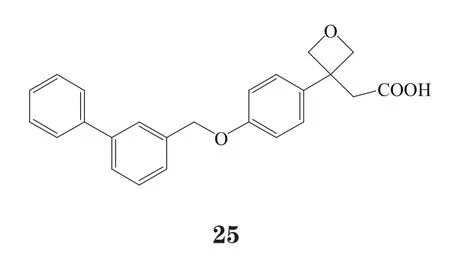
有研究者在对羟基苯丙酸衍生物的修饰改造中,在苯丙酸的羧基和苯环之间引入一个环状结构,结果发现一系列二苯基醚类活性化合物,其中代表性化合物26和27对GPR40的pEC50在7~9之间[37-39]。
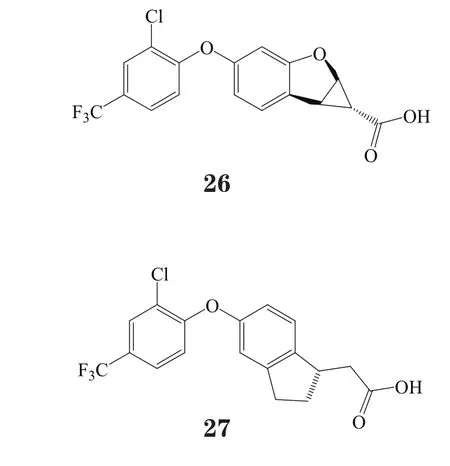
而将不同的苄基基团和对羟基苯丙酸基团相连而形成醚结构,又得到新的一类活性化合物。其中,化合物28对GPR40的pEC50大于8[40],化合物29则是由一个邻甲基苄基基团与对羟基苯丙酸基团相连而成,其目前已进入临床前研究阶段[41]。
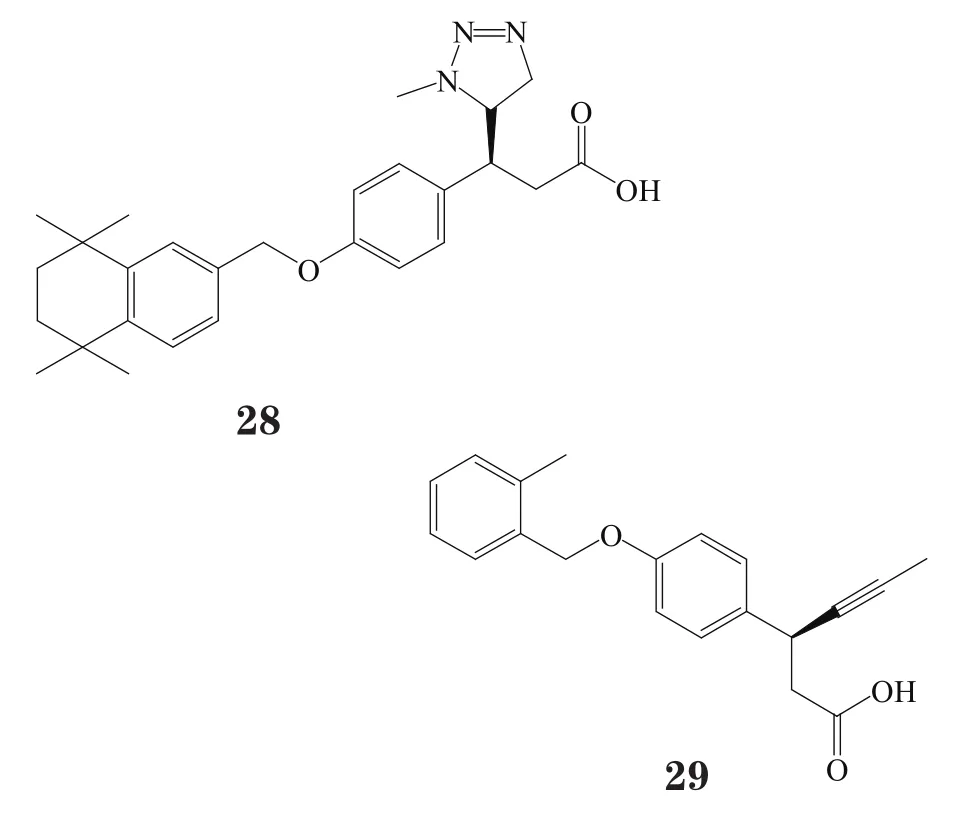
此外,一系列含四氮唑或环丙基的对羟基苯丙酸衍生物类GPR40激动剂也被发掘,如化合物30和AS2575959(31)。在小鼠中进行的一系列药理实验发现,AS2575959能降低糖化血红蛋白浓度而不增加体质量以及食物的摄入,且与DPP-4抑制剂西格列汀联用时,其可协同促进葡萄糖依赖性胰岛素分泌和血浆GLP-1分泌[42]。
近期,对羟基苯丙酸衍生物类GPR40激动剂中苯丙酸结构的修饰改造研究有了重大突破,一系列苯丙酸结构的生物电子等排体被发现和应用,其中,通过高通量筛选发现的新型3,5-二氧-1,2,4- 二唑烷类化合物具有血糖依赖性降糖作用,因此无低血糖风险,如代表性化合物32[42]。

2.3 其他类型化合物
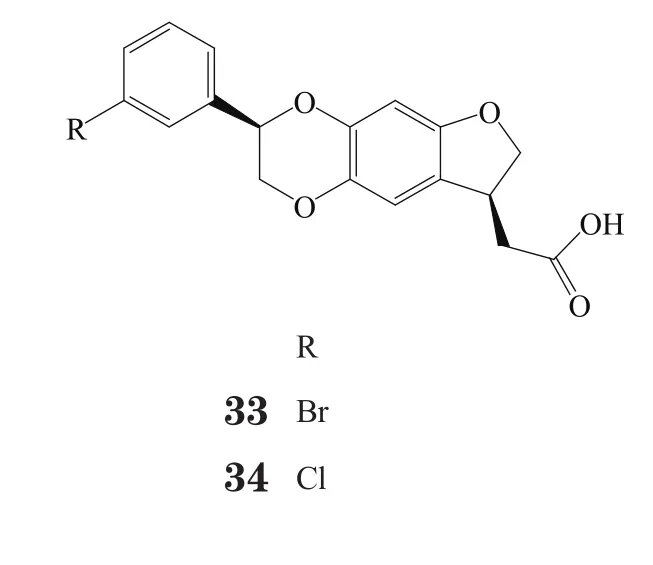
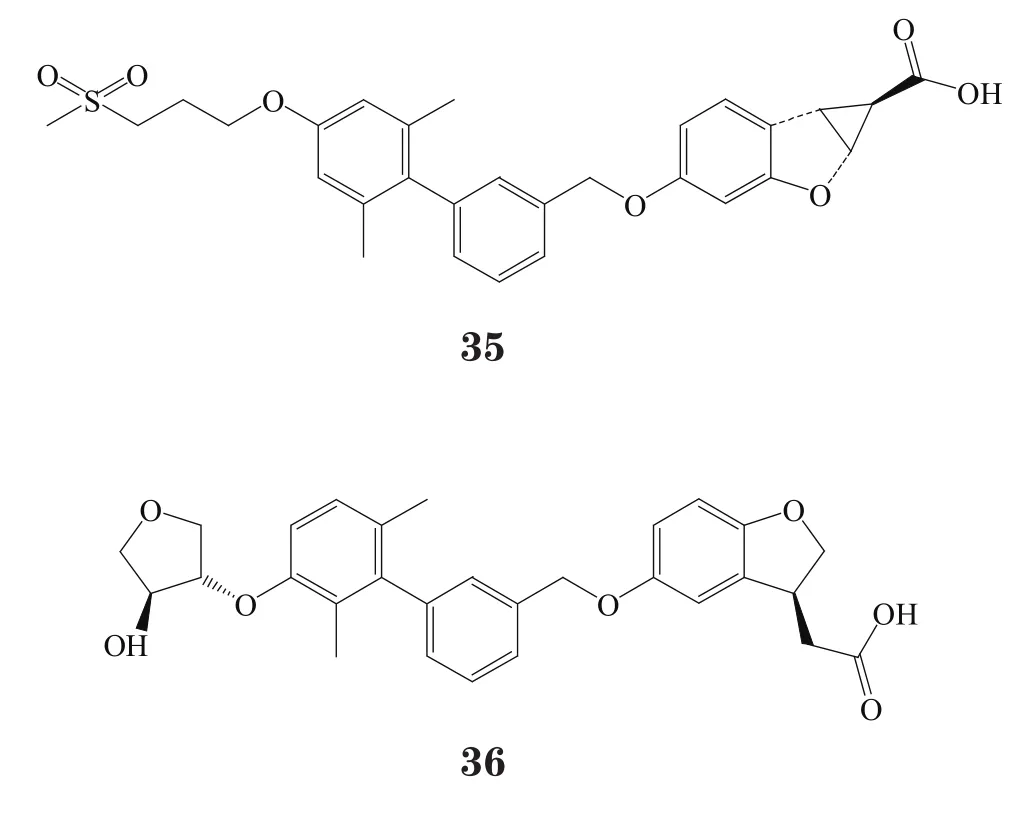
恒瑞公司发现了一系列与武田公司开发的GPR40激动剂结构相类似的化合物,其代表性化合物33~36对GPR40的EC50分别为14、34、26和41 nmol·L-1。恒河猴体内实验表明,化合物33和34具有良好的药理作用,是强效GPR40激动剂。但在小鼠和恒河猴的体内试验中,化合物33和34的活性呈现物种差异性[43-45]。
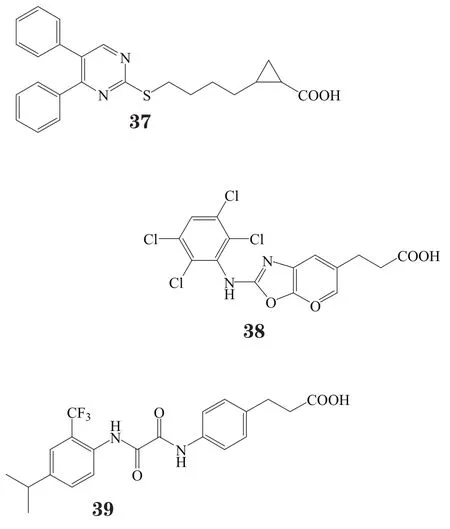
赛诺菲公司则发现一些另类结构的GPR40激动剂,如含烷基羧酸的嘧啶类衍生物(代表性化合物37)和苯并 唑类化合物(代表性化合物38)[46],以及进一步将 唑环变构为乙二酰胺结构而得到的一系列化合物(代表性化合物39)和在苯丙酸β位引入取代基以防止β氧化而得到的化合物40~42。对人GPR40受体进行体外实验时,通过测定胰岛素信号通路中三磷酸肌醇的含量来监测胰岛素的分泌,结果发现,化合物41(SAR1)能显著促进胰岛素分泌。在雌性ZDF大鼠中进行的口服糖耐量实验显示,SAR1的最小有效剂量是1 mg·kg-1[47]。
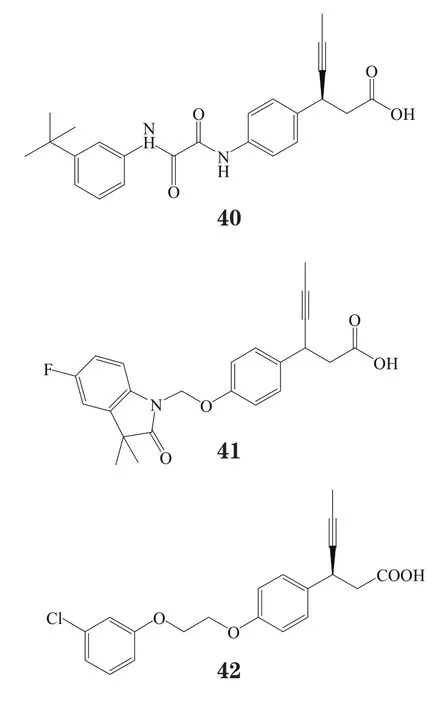
2.4 亲水性改造
一系列合成的小分子GPR40激动剂已见诸多文献报道,其中TAK-875 和AMG-837均已进入临床研究阶段。然而,这些化合物存在的一个普遍问题是都具有相对较高的亲脂性,这是由于内源性GPR40激动剂为FFA,因此要求小分子GPR40激动剂疏水性较高,可是这一性质导致了这些小分子GPR40激动剂化合物表现出较差的药代动力学性质和代谢稳定性以及产生细胞毒性与脱靶效应等[48]。研究表明,这类化合物的ClogP值不应超过4[15]。因而,在小分子GPR40激动剂化合物的设计和修饰改造时,应特别注意配体-亲脂性效率(LLE),在维持化合物活性不变的同时,对其进行亲水性改造,以降低脂毒性。
Negoro等[16]在研究中发现化合物43对GPR40具有良好的激动活性(EC50=30 nmol·L-1),但其亲脂性较高(LogD=4.19),过高的亲脂性使它可能与多种受体结合而产生细胞毒性。因此,该研究小组尝试在化合物43结构中寻找可引入亲水基团的合适位点,以降低其细胞毒性。结果,发现了一系列4-烷氧基联苯衍生物,并发现在2,4-二甲基联苯部分的4位引入亲水性基团,可在保持甚至提高化合物活性的同时降低其亲脂性。其中一些化合物实现了细胞毒性和药代动力学性质的平衡,如化合物44含有一个磺基,在10 nmol·L-1时无细胞毒性,但在30 nmol·L-1时仍具有细胞毒性,而具相似结构的化合物45其LogD为 2.78,在30 nmol·L-1时细胞毒性明显降低。此外,在苯丙酸与二甲基联苯的连接臂上,氧原子被氮原子替代,可更好地降低化合物的细胞毒性,如代表性化合物46对GPR40有较好的激动活性(EC50=29 nmol·L-1),LogD为2.14,在30 nmol·L-1时无细胞毒性,同时由于亲水性的改善,在小鼠口服糖耐量实验中,其可在更低剂量(0.3 mg·kg-1)下显著提高降血糖效果。
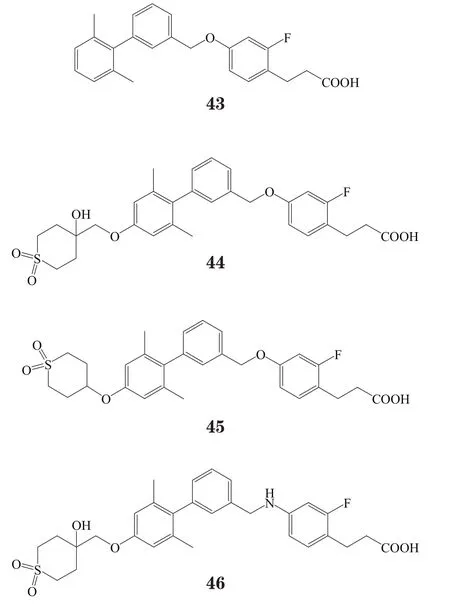
3 结语
GPR,特别是GPR40作为治疗2 型糖尿病的新靶点,在胰岛素分泌通路的上游有调节血糖的能力,因此,GPR40 激动剂可增强胰岛β细胞功能,促进胰岛素的释放增加,提高机体对胰岛素的敏感性,从而达到治疗糖尿病的效果。作为2型糖尿病的治疗靶点,GPR40具有多种潜在优势:第一,由于GPR40是葡萄糖依赖性地调节胰岛素分泌,故其激动剂不会或者很少导致低血糖危险,且可能比其他类胰岛素分泌促进剂(如磺酰脲类和格列奈类)更具疗效;第二,GPR40在体内的分布比较有限,因此其激动剂可能较少产生副作用;第三,GPR40能调节GLP-1释放,所以其激动剂有助于控制摄食、减轻体质量以及减少β细胞凋亡,与其他途径治疗药物有着异曲同工之妙。
然而,GPR40激动剂目前还处于开发的初期,其部分药理性质及临床不良反应(如TAK-875所致肝毒性)机制等尚未明确。因此,GPR40用作抗糖尿病药物靶点,尚有极大的研究空间。
[1]Wild S, Roglic G, Green A, et al.Global prevalence of diabetes: estimates for the year 2000 and projections for 2030[J].Diabetes Care, 2004, 27(5): 1047-1053.
[2]Vassilatis D K, Hohmann J G, Zeng H, et al.The G protein-coupled receptor repertoires of human and mouse[J].Proc Natl Acad Sci U S A, 2003, 100(8): 4903-4908.
[3]Ellis C, Smith A.Highlighting the pitfalls and possibilities of drug research[J].Nat Rev Drug Discov, 2004, 3(3): 238-278.
[4]Kebede M A, Alquier T, Latour M G, et al.Lipid receptors and islet function: therapeutic implications?[J].Diabetes Obes Metab, 2009, 11 (Suppl 4): 10-20.
[5]Insel P A, Tang C M, Hahntow I, et al.Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets[J].Biochim Biophys Acta, 2007, 1768(4): 994-1005.
[6]Schmitz G, Ecker J.The opposing effects of n-3 and n-6 fatty acids [J].Prog Lipid Res, 2008, 47(2): 147-155.
[7]Miyauchi S, Hirasawa A, Ichimura A, et al.New frontiers in gut nutrient sensor research: free fatty acid sensing in the gastrointestinal tract[J].J Pharmacol Sci, 2010, 112(1): 19-24.
[8]Sauer L A, Dauchy R T, Blask D E.Mechanism for the antitumor and anticachectic effects of n-3 fatty acids[J].Cancer Res, 2000, 60(18): 5289-5295.
[9]Louet J F, Chatelain F, Decaux J F, et al.Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I) expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-independent pathway[J].Biochem J, 2001, 354(Pt 1): 189-197.
[10]Xiong Y, Miyamoto N, Shibata K, et al.Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41[J].Proc Natl Acad Sci U S A, 2004, 101(4): 1045-1050.
[11]Shapiro H, Shachar S, Sekler I, et al.Role of GPR40 in fatty acid action on the beta cell line INS-1E[J].Biochem Biophys Res Commun, 2005, 335(1): 97-104.
[12]Ubink-Veltmaat L J, Damoiseaux R A, Rischen R O, et al.Please,let my doctor be obese: associations between the characteristics of general practitioners and their patients with type 2 diabetes[J].Diabetes Care, 2004, 27(10): 2560.
[13]Fujiwara K, Maekawa F, Yada T.Oleic acid interacts with GPR40 to induce Ca2+signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+channel and link to insulin release[J].Am J Physiol Endocrinol Metab, 2005, 289(4): E670-E677.
[14]McKeown S C, Corbett D F, Goetz A S, et al.Solid phase synthesis and SAR of small molecule agonists for the GPR40 receptor[J].Bioorg Med Chem Lett, 2007, 17(6): 1584-1589.
[15]Burant C F.Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes[J].Diabetes Care, 2013, 36 (Suppl 2): S175-S179.
[16]Negoro N, Sasaki S, Mikami S, et al.Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist[J].ACS Med Chem Lett, 2010, 1(6): 290-294.
[17]Takeda.Comparison of TAK-875 with sitagliptin when used in combination with metformin in patients with type 2 diabetes[EB/OL].[2014-10-27].http://clinicaltrials.gov/ct2/show/NCT01834274,2012.
[18]Takeda.Effcacy and safety of TAK-875 compared to glimepiride when used with metformin in participants with type 2 diabetes[EB/OL].[2014-10-27].http://clinicaltrials.gov/show/NCT01481116.2012.
[19]Takeda.TAK-875 (fasiglifam) in combination with sitagliptin in adults with type 2 diabetes[EB/OL].[2014-10-27].http://clinicaltrials.gov/ show/NCT01829464.2013.
[20]Srivastava A, Yano J, Hirozane Y, et al.High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875[J].Nature, 2014, 513(7516): 124-127.
[21]Brown S P, Dransfield P J, Vimolratana M, et al.Discovery of AM-1638: a potent and orally bioavailable GPR40/FFA1 full agonist[J].ACS Med Chem Lett, 2012, 3(9): 726-730.
[22]Yasuma T N N, Fukatsu K.Condensed ring compound: US, 10/558846[P].2006-11-16.
[23]Reifel Miller A, Cox A, Briere D, et al.In vitro insulin action[C]//González Brao M.Proceedings of the 48th EASD.Berlin: Thomson Reuters, 2012.
[24]Reifel Miller A, Cox A, Briere D, et al.In vitro insulin action[C]//American Diabetes Association.Proceedings of 72nd American Diabetes Association.Philadelphia: American Diabetes Association, 2012.
[25]Hamdouchi C.A novel 1,2,3,4-tetrahydroquinoline derivative useful for the treatment of diabetes: US, 2012/050051[P].2013-02-21.
[26]Christiansen E, Hansen S V, Urban C, et al.Discovery of TUG-770: a highly potent free fatty acid receptor 1 (FFA1/GPR40) agonist for treatment of type 2 diabetes[J].ACS Med Chem Lett, 2013, 4(5): 441-445.
[27]Kebede M, Alquier T, Latour M G, et al.The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding[J].Diabetes, 2008, 57(9): 2432-2437.
[28]Poitout V, Lin D C.Modulating GPR40: therapeutic promise and potential in diabetes[J].Drug Discov Today, 2013, 18(23/24): 1301-1308.
[29]Mancini A D, Poitout V.The fatty acid receptor FFA1/GPR40 a decade later: how much do we know?[J].Trends Endocrinol Metab, 2013, 24(8): 398-407.
[30]Ferdaoussi M, Bergeron V, Zarrouki B, et al.G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1[J].Diabetologia, 2012, 55(10): 2682-2692.
[31]Lin D C, Zhang J, Zhuang R, et al.AMG 837: a novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents[J].PloS One, 2011, 6(11): e27270.
[32]Tobacco J.A phase 2 study of JTT-851[EB/OL].[2014-11-07].http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicC TI-121730.2011.
[33]Tobacco J.Safety and effcacy study of JTT-851 in patients with type 2 diabetes mellitus[EB/OL].[2014-11-07].http://clinicaltrials.gov/show/ NCT01699737.2011.
[34]Shimada T U H, Tsutsumi K, Aoyagi K, et al.Spiro-ring compound and use thereof for medical purposes: JP, 2008069296[P].2009-04-30.
[35]Piramal.Piramal receives US FDA approval for anti-diabetic molecule, GPR40 agonist P11187[EB/OL].[2014-11-17].http://www.pharmabiz.com/NewsDetails.aspx?aid=75330&sid=2.2011.
[36]Piramal.Determination of safety,tolerability,pharmacokinetics,food effect& pharmacodynamics of single & multiple doses of P11187[EB/ OL].[2014-11-17].http://clinicaltrials.gov/show/NCT01874366.2011.
[37]Kuo G H, Song F, Gunnet J, et al.GPR40 agonists: US, 11/939039[P].2008-07-24.
[38]Min G E, Jiafang H E, Lau F W Y, et al.Antidiabetic bicyclic compounds: WO, 2007136572[P].2007-11-29.
[39]Min G E, Lin S, Walsh S, et al.Antidiabetic bicyclic compounds: WO, 2008054675[P].2008-05-08.
[40]Brown S, Dransfeld P, Houze JB, et al.Benzo-fused compounds for use in treating metabolic disorders: WO, 2008030618[P].2008-08-28.
[41]Akerman M, Houze J, Lin D C H, et al.Compounds, pharmaceutical compositions and methods for use in treating metabolic disorders: US, 2005/005815[P].2005-09-22.
[42]Negoro K O K, Yonetoku Y, Kuramoto K, et al.Tetrazole compound: JP, 2010/057035[P].2010-10-28.
[43]Lu H, Fei H, Yang F, et al.Discovery of novel orally bioavailable GPR40 agonists[J].Bioorg Med Chem Lett, 2013, 23(10): 2920-2924
[44]吕贺军, 董庆, 费鸿博, 等.苯并二氧六环类衍生物、其制备方法及其在医药上的应用: 中国, 201210300982.7[P].2012-08-22.
[45]杨芳龙, 范江, 董正, 等.稠合环类衍生物、其制备方法及其在医药上的应用: 中国, 201210351802.8[P].2012-09-19.
[46]Kang H.Use of the fetal reprogramming of a PPAR agonist: WO, 2012/030165 [P].2011-03-08.
[47]Himmelsbach F B R, Eckhardt M, Hamprecht D, et al.Antidiabetic bycyclic compounds: WO, 2014/130608[P].2014-06-08.
[48]Gleeson M P.Generation of a set of simple, interpretable ADMET rules of thumb [J].J Med Chem, 2008, 51(4): 817-834.
Recent Research Advances in G-protein Coupled Receptor 40
Agonists as Therapeutic Agents for Type 2 Diabetes
ZHOU Muxing, YANG Lingyun, ZHOU Jinpei, ZHANG Huibin
(Jiangsu Key Laboratory of Anti-metabolic Drugs, China Pharmaceutical University, Nanjing 210009, China)
Type 2 diabetes accounts for about 90% of all cases of diabetes.Novel therapeutic drugs for type 2 diabetes in current research and development control blood glucose level mainly by regulating glycometabolism pathways, which could strengthen the function of islet beta cells, promote insulin secretion and improve the body's sensitivity to insulin through activating G-protein coupled receptors(GPRs) , especially GPR40, so as to achieve the goal of treating diabetes.GPR40 as novel therapeutic targets for type 2 diabetes, with its potential advantages, has attracted much attention in the feld of diabetes treatment.GPRs and Their ligands——free fatty acids were Introduced.The research progress on GPR40 agonists with different structures was especially reviewed.
GPR40; agonist; type 2 diabetes; insulin secretion; blood glucose level
R962; R977.15
A
1001-5094(2015)01-0013-10
接受日期:2014-12-03
项目资助:国家“重大新药创制”科技重大专项(No.2013ZX09301303-002); 江苏省自然科学基金(No.BK 20141349)
*通讯作者:张惠斌, 研究员;
研究方向: 心血管疾病与肿瘤治疗药物;
Tel:025-83271302;E-mail:zhanghb80@163.com
