不对称树枝状咔唑类衍生物的合成
2014-12-31刘课艳董雪芬王彭敏石先莹
偶 辉,刘课艳,董雪芬,王彭敏,石先莹
(陕西师范大学 化学化工学院,陕西省大分子科学重点实验室,陕西 西安 710119)
树状大分子是近年来国内外开发的一类新型功能高分子,结构上具有高度几何对称性的特点.它由各种初始核构筑而成,其外围支化层(代数)呈几何级数增长,最外层的末端基团分布在树状大分子表面.树状大分子因其特殊的结构性,在纳米复合材料、催化剂、膜材料、废水处理、高分子材料等方面已显示出广阔的应用前景.在过去的20年里,构建树枝状分子引起了研究者们的广泛关注[1-3].
含有咔唑单元的化合物是一类具有独特性质的分子,不仅具有咔唑单元高效的光学和电学性质,也是一种高效的空穴传输材料[4-10]和主体发光材料[11-14].在树状高分子的研究中,树枝状咔唑类衍生物是一类结构新颖、潜在应用价值高的材料,目前树状咔唑分子已用于纳米复合材料的研制和树枝状有机发光二极管的发射器的制造.
多数树枝状咔唑衍生物都属于N-芳基咔唑类化合物,此类化合物合成方法主要是利用Ullmann反应、Buchwald-Hartwig偶联反应形成C—N键(图1).但是传统的这两类反应应用在树枝状咔唑衍生物的合成中,存在反应时间长、产率低等缺点.
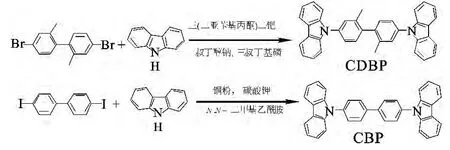
图1 乌尔曼偶联反应和布赫瓦尔德-哈特维希反应Fig.1 Ullmann and Buchwald-Hartwig coupling reaction
有关树状咔唑类衍生物的合成,以1,4-双咔唑基苯和1,8-双咔唑基联苯为中心的树枝状咔唑衍生物报道很少.2004年,虽然王利祥[15]小组以1,4-双咔唑基苯为基本骨架,通过外围接枝不同数量的二苯胺为树枝结构,得到了一系列空穴传输材料;这些化合物热稳定性高,玻璃化温度(Tg)范围在141~157℃,是一类性能较好的空穴传输材料;但是此类外围结构为咔唑基团构筑的树枝状化合物却未见报道.
本文以1,4-二溴苯和1,8-二碘联苯为原料,分别与咔唑发生乌尔曼反应得到两种成对偶联分子.在发散法设计思想的导向下,以此结构为中心导入了不同数量的咔唑,从而合成了3种新颖的树枝状咔唑类衍生物.
1 实验
1.1 主要仪器与试剂
超导傅里叶数字化核磁共振仪(瑞士Bruker公司AVANCE 300MHz,400MHz),基质辅助激光解吸电离飞行时间质谱仪(MS(MALDI-TOF)瑞士Bruker公司).
咔唑、对二溴苯、18-冠-6均购置于上海达瑞精细化学品有限公司,无水碳酸钾、N-溴代丁二酰亚胺(NBS)、碘化亚铜、碘化钾、一水合邻菲罗啉均购于国药集团化学试剂有限公司,N,N-二甲基乙酰胺(DMAc氢化钙干燥处理),N,N-二甲基甲酰胺(DMF氢化钙干燥处理),二碘联苯(根据文献[16]自制).
1.2 合成与表征
树枝状咔唑类衍生物的合成路线如图2所示.
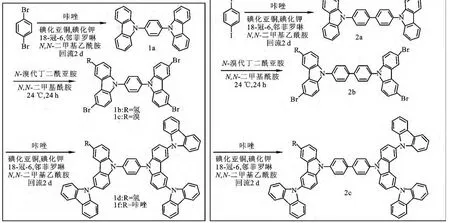
图2 合成路线Fig.2 The synthesis course
1.2.1 中间体1,4-双咔唑基苯(1a)的合成 将对二溴苯(7.08g,30.27mmol),咔唑(15.06g,90.17mmol),碳酸钾(25.00g,181.00mmol),碘化亚铜(4.20g,30.00mmol),18-冠-6(0.79g,3.00mmol),一 水 合 邻 菲 罗 啉 (0.59g,3.00 mmol),碘化钾(0.05g,0.30mmol)混合后加入DMAc 250mL,氮气保护下回流48h,冷却至室温后,缓慢倒入2L冰水混合液中,搅拌1h,过滤得到固体,分别用二氯甲烷100mL萃取5次,有机相用饱和食盐水洗涤3次,无水硫酸镁干燥过夜,旋转蒸发得到粗产品,柱色谱分离(PE∶DCM=10∶1~4∶1)得到7.34g棕色固体1a,收率70%.1H NMR(300MHz,CDCl3)δ:8.18(d,J=7.6 Hz,4H),7.80(s,4H),7.65~7.40(m,8H),7.40~7.27(m,4H);13CNMR (75MHz,CDCl3)δ:140.7,136.7,128.4,126.0,123.7,120.5,120.3,109.8.MS(MALDI-TOF):实验值m/z[M]+408.036(计算值:C30H20N2,408.163).
1.2.2 多溴取代物1b、1c的合成 将1a(0.40g,1.00mmol),溶于100mLDMF中,避光,于冰浴中缓慢滴加10mL含 NBS(0.71g,4.00mmol)的DMF溶液,滴加完毕后撤去冰水浴,将装置改为油浴,缓慢升温至40℃,保持此温度反应12h,生成的沉淀过滤收集,依次用DMF、丙酮、蒸馏水、乙醇洗涤沉淀3次,干燥得到0.62g白色固体.
由于产物不能溶于常用溶剂,未能得到核磁数据.通过MS(MALDI-TOF)检测说明此化合物是一个混合物,主要包含3,3’,6-三溴-1,4-双咔唑基苯(1b)和3,3’,6,6’-四溴-1,4-双咔唑基苯(1c).
1bMS (MALDI-TOF):实 验 值:m/z[M]+641.522,[M+2]+643.487,[M+4]+645.524(计算值:C30H17Br3N2,[M]+641.891,[M+2]+643.901,[M+4]+645.899).
1cMS (MALDI-TOF):实验值:m/z[M]+719.534,[M+2]+721.526,[M+4]+723.484,[M+6]+725.527,[M+8]+727.547(计算值:C30H16Br4N2,[M]+719.805,[M+2]+721.803,[M+4]+723.801,[M+6]+725.799,[M+8]+727.805).
鉴于此类化合物溶解度较差,对此类化合物无需纯化直接用于下一步反应.
1.2.3 树枝状咔唑衍生物1d、1f的合成 将多溴取代物1b、1c的混合物(0.72g,1.00mmol)加入到100mL 三口烧瓶中,咔唑 (0.68g ,4.00 mmol),一水合邻菲罗啉(0.02g,0.10mmol),碘化钾(0.01g,0.10mmol)碳酸钾(0.50g,4.00 mmol),碘化亚铜(0.02g,0.10mmol),加入100 mL DMAc中,在氮气保护下充分搅拌30min,缓慢升温至回流,保持此温度48h.冷却至室温,抽滤得到滤液,滤液倒入500mL冰水中,搅拌30 min,过滤得到固体,依次用氨水、蒸馏水、氨水洗涤3次,二氯甲烷100mL萃取3次,有机相用蒸馏水洗涤3次,无水硫酸镁干燥,旋转蒸发得到粗产品,柱色谱分(PE∶DCM=3∶1~1∶10),得到0.43g白色固体1d和0.01g黄色固体1f,产率1d 40%、1f1%.核磁、质谱数据如下:
1d1H NMR (400MHz,CDCl3)δ:8.16(s,3H),7.98(d,J=7.4Hz,7H),7.73(s,4H),7.69~7.34(m,9H),7.25(s,10H),7.12(d,J=7.8Hz,8H).13CNMR (100MHz,CDCl3)δ:141.9,141.8,141.5,140.6,139.9,137.2,136.5,130.9,130.5,128.9,128.8,128.7,127.0,126.5,126.0,125.8,124.8,124.3,123.3,121.0,120.9,120.4,120.0,119.9,119.8,113.3,110.9,110.1,109.8,109.7.MS(MALDI-TOF):实验值m/z[M]+903.226 (计算值:C66H41N5,903.336).
1f1H NMR(400MHz,CDCl3)δ:8.27~8.24(d,J=4.1Hz,4H),8.14~8.09(m,9H),7.94~7.88(m,4H),7.81~7.76(m,4H),7.65~7.63(d,J=3.3Hz,6H),7.45~7.36 (m,14H),7.24~7.23 (d,J=3.6Hz,7H);13C NMR(100MHz,CDCl3)δ:141.8,140.5,130.9,128.9,128.8,128.7,126.6,126.0,124.3,123.3,120.4,119.8,111.3,109.7.MS(MALDI-TO F):m/z[M]+1 068.681(计 算 值:C78H48N6,1 068.394).
1.2.4 中间体1,8-双咔唑基联苯(2a)的合成 氮气保护下将1,8-二碘联苯(2.00g,4.92mmol),咔唑(1.80g,10.00mmol),碳酸钾(5.44g,39.40mmol),碘化亚铜(0.38g,2.00mmol),碘化钾(0.01g,0.49mmol),一水合邻菲罗啉(0.19 g,1mmol),18-冠-6(0.26g,0.98mmol),混合后加入100mL DMAc,室温搅拌30min,缓慢升温至回流,保持此温度24h,冷却至室温,抽滤,滤液倒入500mL蒸馏水中,搅拌30min,析出的固体通过过滤收集,依次用20mL氨水、蒸馏水洗涤3次,二氯甲烷100mL萃取3次,有机相分别用饱和食盐水、蒸馏水洗涤3次,无水硫酸钠干燥过夜,旋转蒸发浓缩得到粗产品,柱色谱分离(PE∶DCM=10∶1~3∶1)得到2.50g淡黄色固体2a.产率80%.1H NMR (400MHz,CDCl3)δ:8.03(dd,J=2.7,7.6Hz,4H),7.87~7.70(m,4H),7.60(dd,J=6.4,2.0Hz,4H),7.47~7.29 (m,8H),7.22 (t,J=7.4Hz,4H);13C NMR(100MHz,CDCl3)δ:139.8,138.2,136.2,127.5,126.4,125.0,122.5,119.3,119.1,108.8.MS(MALDI-TOF):实 验 值m/z[M]+484.121(计算值:C36H24N2,484.194).
1.2.5 多溴取代物2b 称取1,8-双咔唑基联苯(2a)(0.48g,1.00mmol),溶于100mL DMF中,避光,于冰浴中缓慢滴加10mL含NBS(0.71g,4.00mmol)的DMF溶液,滴加完毕后撤去冰水浴,将装置改为油浴,缓慢升温至40℃,保持此温度搅拌12h,生成的沉淀通过过滤收集,依次用DMF、丙酮、蒸馏水、乙醇洗涤沉淀3次,干燥得到0.65g白色固体2b,由于该产品不能溶解常用溶剂,处理完毕无须纯化直接用于下一步反应.
1.2.6 树枝状咔唑衍生物2c的合成 氮气保护下将2b产物(0.80g,1.00mmol)加入到100mL 三口烧瓶中,咔唑(0.68g,4mmol),碳酸钾(0.50 g,4.00mmol),一水合邻菲罗啉(0.02g,0.10 mmol),碘化亚铜(0.02g,0.10mmol),碘化钾(0.01g,0.10mmol),加入100mL DMAc,搅拌30min,缓慢升温至回流,保持此温度24h,冷却至室温,抽滤得到滤液,滤液倒入500mL冰水中,搅拌30min,抽滤得到固体,依次用氨水、蒸馏水、氨水洗涤3次,二氯甲烷100mL萃取3次,有机相用蒸馏水洗涤3次,无水硫酸镁干燥,旋转蒸发有机相得到粗产品,柱色谱分离(PE∶DCM=3∶1~1∶2)得到0.57g白色固体2c,产率50%.
1H NMR(400MHz,CDCl3)δ:8.26~7.97(m,10H),7.89~7.74 (m,4H),7.72~7.42(m,11H),7.29(d,J=7.3Hz,13H),7.17(d,J=8.0Hz,7H).13CNMR (100MHz,CDCl3)δ:140.8,140.7,140.6,140.4,139.5,139.1,138.9,138.6,138.3,136.0,135.7,129.5,128.4,127.8,127.7,127.6,126.5,126.4,125.7,125.3,125.0,124.9,124.5,123.8,123.5,123.0,122.4,122.2,122.1,119.6,119.4,118.7,118.6,112.3,110.6,110.3,110.2,109.9,109.1,108.7,108.6.MS(MALDI-TOF):实 验 值m/z[M]+979.452(计 算值:C72H45N5,979.367).
2 结果与讨论
在利用乌尔曼偶联反应合成1,4-双咔唑基苯时,我们对文献报道的合成方法进行了改进[15,17].添加适当的配体一水合邻菲罗啉和助剂碘化钾后,反应时间明显缩短(由文献报道的7d缩短为2d),反应收率显著提高(从文献报道的50%提高到70%),为下一步的合成奠定了良好的基础.其中添加一水合邻菲罗啉的作用可能是它和铜(I)盐形成配合物,使生成的中间体更为稳定,从而提高了产率;助剂碘化钾增加碘化亚铜在溶剂中的溶解度,有效促进反应的进行.
NBS作为溴化剂与1a、2a发生溴化时我们通过质谱MS(MALDI-TOF)检测看出主要生成的是多溴代产物1b、1c、2b,这些物质不溶于常见溶剂,造成纯化的困难,但利用混合的多溴代化合物进一步发生乌尔曼反应合成树状咔唑衍生物时,不影响目标产物的合成.
实验过程中利用质谱监测(MS(MALDI-TOF)发现多溴代产物在与咔唑发生乌尔曼偶联反应,未加配体时反应极为困难,甚至无反应,当添加配体一水合邻菲罗啉和助剂碘化钾后,能以较高收率制得目标产物1d、2c.
3 结论
本文通过改进传统的乌尔曼偶联反应成功合成了3种新型树枝状咔唑类衍生物,同时运用核磁(1H NMR,13C NMR)、MS(MALDI-TOF)对3种结构新颖的树枝状咔唑衍生物进行了结构表征.在乌尔曼偶联反应中,采用低毒廉价的铜催化剂得到了良好的反应收率,避免了昂贵钯催化剂的使用.本研究丰富了树状咔唑类衍生物的种类,也为这类树状高分子化合物提供了一条简单、高效的合成方法.
[1]Astruc D,Boisselier E,Ornelas C.Dendrimers designed for functions:from physical,photophysical,and supramolecular properties to applications in sensing,catalysis,molecular electronics,photonics,and nanomedicine[J].Chemical Reviews,2010,110:1857-1959.
[2]Caminade A M,Majoral J P.Dendrimers and nanotubes:a fruitful association[J].Chemical Society Reviews,2010,39:2034-2047.
[3]Franc G,Kakkar A K.“Click”methodologies:efficient,simple and greener routes to design dendrimers[J].Chemical Society Reviews,2010,39:1536-1544.
[4]Du Pa,Zhu Weihong,He Tian,et al.Dendron-functionalized macromolecules:enhancing core luminescence and tuning carrier injection[J].Macromolecules,2004,37:4387-4398.
[5]Kimoto A,Cho J S,Higuchi M,et al.Novel carbazole dendrimers having a metal coordination site as a unique hole-transport material[J].Macromolecular Symposium,2004,209:51-56.
[6]Kimoto A,Miyake T,Yamamoto K M.Novel holetransport material for efficient polymer light-emitting diodes by photoreaction[J].Macromolecular Rapid Communication,2005,26:597-601.
[7]Albrecht K,Yamamoto K.Organic light-emitting diodes with novel carbazole dendrimer as a photocrosslinking hole-transfer layer[J].Journal of Photopolymer Science and Technology,2006,19:175-176.
[8]Promarak V,Saengsuwan S,Keawin T.Synthesis and properties of stable amorphous hole-transporting molecules for electroluminescent devices[J].Tetrahedron Letters,2006,47:8949-8952.
[9]Promarak V,Ichikawa M,Sudyoadsuk T.Synthesis of electrochemically and thermally stable amorphous holetransporting carbazole dendronized fluorene[J].Synthetic Metals,2007,157:17-22.
[10]Albrecht K,Kasai Y,Kimoto A,et al.The synthesis and properties of carbazole-phenylazomethine double layer-type dendrimers[J].Macromolecules,2008,41:3793-3800.
[11]Tsai M H,Hong Y H,Chang C H,et al.3-(9-Carbazolyl)carbazoles and 3,6-di(9-carbazolyl)carbazoles as effective host materials for efficient blue organic electrophosphorescence[J].Advanced Materlals,2007,19:862-866.
[12]Xie Zhiyuan,Jing Xiabin,Wang Lixiang,et al.Solu tion-processable carbazole-based conjugated dendritic hosts for power-efficient blue-electrophosphorescent devices [J]. Advanced Materlals, 2009, 21:4983-4986.
[13]Yang Jun,Ye Tengling,Ma Dongge,et al.“Click”synthesis of a bipolar dendrimer as a host material for electrophosphorescent devices[J]. Macromolecular Chemistry and Physics,2010,211:1969-1976.
[14]Soh M S,Santamaria S A G,Williams E L,et al.Solution processable high band gap hosts based on carbazole functionalized cyclic phosphazene cores for application in organic light-emitting diodes [J].Journal of Polymer Science Part B:Polymer Physics,2011,49:531-539.
[15]Zhang Qian,Chen Jiangshan,Wang Lixiang,et al.Novel hole-transporting materials based on 1,4-bis(carbazoly)benzene for organic light-emitting devices[J].Journal of Materials Chemistry,2004,14:895-900.
[16]Li Zhonghui,Wong Manshing,Tao Ye,et al.Synthesis and functional properties of strongly luminescent diphenylamino end-capped oligophenylenes [J]. The Journal of Organic Chemistry,2004,69:921-927.
