甲基环己烷催化裂化分子水平反应动力学模型的建立
2014-12-31郭锦标王鑫磊
张 旭,郭锦标,周 祥,王鑫磊,于 博,付 翁
(中国石化 石油化工科学研究院,北京 100083)
当前,环保法规不但要求减少空气污染物排放量,而且对燃油产品的各项质量指标提出了新的要求。例如,欧Ⅴ标准规定清洁汽油和柴油的硫质量分数上限值为10μg/g,对苯和整体芳烃含量也作出了明确的限制[1],表明新的环保法规对车用燃油的质量标准提出了更高的要求。另外,油品重质化劣质化加重的趋势也是不争的事实。目前主要是将重质油加氢处理后再进行催化裂化处理,生产清洁汽、柴油,这一加工过程首先是将芳烃变成环烷烃,再得到清洁汽、柴油。炼油工业经历了150多年的发展历史,现在较难实现重大的技术创新。为了更高效地利用石油资源,应将对石油及其转化规律的认识上升到分子水平。这就要求动力学模型的发展要跟上这个新的要求,对复杂反应体系中产物分布及其性质作出更细致、准确的预测,以反映出其与原料性质、操作变量、催化剂之间的内在关联。
催化裂化反应动力学模型经历了关联模型[2]、集总动力学模型[3-5]到组分划分更为精细的分子水平动力学模型[6-9]的转变。关联模型在早期得到了广泛的应用,集总动力学模型虽然仍是目前指导催化裂化过程的最主要动力学方法,但是没有赋予充分的化学意义,预测能力有限,并且无法提供详细的石油分子组成及结构信息。过去的二十年,分子水平动力学模型开发得到了快速的发展,该模型使人们能够深入认识反应机理,定量预测前所未有的分子细节。
在20世纪80年代就有相关模型化合物的分子水平反应动力学模型研究报道,如烷烃[9-10]、烷基苯[11]的分子水平动力学机理模型。由于烃类化合物的催化裂化反应涉及到大量化学反应,构成了一个复杂的反应网络,这也使得建立催化裂化动力学模型变得十分困难。先后出现了结构化模型(Structural model,SM)、单事件模型(Single event kinetic model,SEKM)、结构导向型集总模型(Structure oriented lumping,SOL)、机理模型等。每一种模型都有其适用范围,在适用范围内具有较好的预测能力。然而,部分模型外推性较差,有的模型需要较深厚的理论基础,建立过程十分复杂,对建模人员的理论知识提出了更高的要求,限制了其运用。因此,需要开发一种操作性强、计算量小、适用范围广的催化裂化动力学模型。
为此,笔者从单环环烷烃入手,以甲基环己烷作为模型化合物,探索建立基于反应路径层面的单环环烷烃催化裂化分子水平反应动力学模型,以期获取一些关于单环环烷烃的定量动力学参数,为建立较完善的单环环烷烃催化裂化分子水平反应动力学模型奠定基础。
1 甲基环己烷催化裂化反应原料和实验数据处理方法
1.1 原料和催化剂
甲基环己烷(MCHA),质量分数99%以上,Alfa Aesar公司产品。2种商业USY分子筛购于Tosoh公司。
取质量分数为50%高岭土、30%USY分子筛以及20%硅溶胶经喷雾干燥、成型后,再通过焙烧、老化处理,得到实验用催化剂,按催化剂平均孔径大小分别表示为 CAT-LC、CAT-SC,其中CAT-LC的孔径较大。
1.2 实验数据处理方法
采用等温固定床反应器进行催化剂活性评价实验[12]。根据建立分子水平动力学模型的要求,在本研究中,采用摩尔流率(mol/s)来表示体系中各组分的浓度。(1)根据原料中各组分的质量分数和密度计算原料的平均密度;(2)根据原料的质量流率计算产物的总质量流率;(3)根据总质量流率和质量分数计算各组分的质量流率;(4)根据各组分的质量流率计算各组分的摩尔流率。
2 建立甲基环己烷催化裂化反应动力学模型的基本步骤
2.1 产物分子分类及命名
虽然MCHA催化裂化反应产物的组成很复杂,但基本上可以将其分为烷烃、烯烃、环烷烃、芳烃4种基本化合物类型。采用简单的烷烃、异构烷烃、烯烃、环烷烃和芳烃(PIONA)分类来表示数量庞大的反应物、中间体和产物,不能满足分子水平动力学模型建立的需要。为了方便后期的反应动力学网络书写,采取了系统命名法的简写形式,如Methane简写为MTA,具体写法见表1。
2.2 动力学参数获取
2.2.1 指前因子
根据过渡态理论、统计热力学和阿伦尼乌斯方程,可得到反应速率常数和指前因子的计算式 (1)和(2)。从过渡态理论出发,经过一定的假设和引入矫正因子,用气相分子的平动熵近似代替难以获得的反应活化态的活化熵与反应物熵差,大大简化了工作量,在一定程度上也保证了数据的准确度[13]。
2.2.2 活化能
采用式(2)可以从理论的角度计算出所有反应的指前因子值,而Ea可以通过量子化学计算软件—Material Studio(MS)软件的DMol3模块计算获得[14]。虽然可以通过模拟计算获得所有反应的Ea,但是催化裂化反应数目众多,其费时较多而不具有现实可操作性。为此,运用反应族的概念来估算每一个基元反应的速率常数。线性自由能关系能够起到热力学和动力学的桥梁作用,Polanyi方程就是其中的一种,将反应的焓变和活化能关联,如式(3)所示。杜梅西克等[15]已经证实Polanyi方程适合于酸催化的正碳离子化学反应。因此每一个反应就可以通过最多3个参数A、E0、α进行描述。
每一个反应的焓变就是产物和反应物之间的焓差,如式(4)所示。对于催化裂化而言,由于反应过程中存在许多正碳离子中间体,不能直接通过实验方式获得,然而量化计算为反应焓变计算提供了条件。目前,基于半经验的VAMP量化计算可以获得反应的生成焓变。将Arrhenius方程和Polanyi方程关联得到的反应速率常数表达式如式(5)所示。
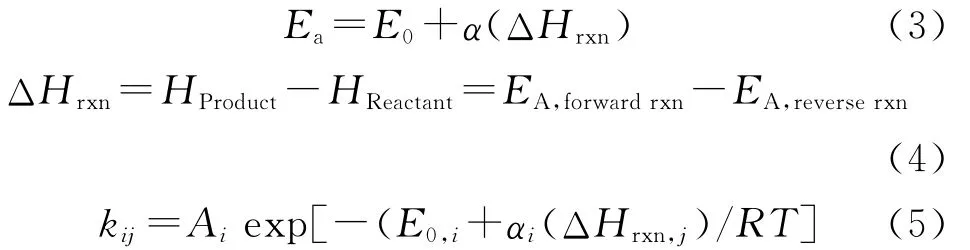
2.3 模型建立与求解
对前面的MCHA催化裂化反应类型进行归类整理,进一步细化反应类型。总的来说,正碳离子主要经历了离子引发、离子转移(变化)、离子消耗3大环节,此外反应过程中有可能结焦,因此构成了一个闭合的反应网络。
以A作为反应物分子,B、C为产物分子进行反应网络的编写。反应网络主要基于路径层面,同时也兼顾机理层面。动力学模型建立过程中主要涉及的方程为式(6)~式(9),它们分别是基元反应速率、反应速率常数、组分浓度及每一类反应的速率常数的表达式。

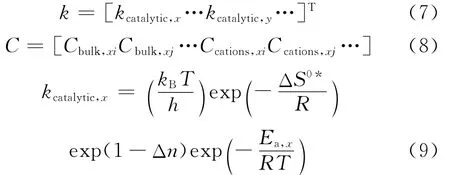
最后,使用对比图的形式列出了组分摩尔流率的预测数据与实验数据对比,可以清晰地看出模型预测结果的准确度,并可得到优化的动力学参数。如果拟合的结果不理想,可以进一步调整算法参数。
3 甲基环己烷催化裂化反应动力学模型的建立
3.1 反应数据处理
MCHA催化裂化反应实验结果表明,MCHA的总转化率随着反应温度的升高和反应时间的延长而提高。在给定的反应时间内,MCHA的转化率会随着反应温度的升高而稳步上升。此外,MCHA的总转化率不受分子筛晶粒内孔扩散阻力影响。在所有反应条件下,采用CAT-LC催化剂比采用CAT-SC催化剂得到的MCHA转化率稍高。
采用GC-MS分析MCHA催化裂化反应产物,检测到了60多种产物。对产物数据进行处理时遵循的原则是,烯烃组分不考虑顺反异构;烷烃组分只区分正构、异构,不考虑支链个数;环烷烃和芳烃只含1个侧链;对于不确定结构的烃类按照异构烃分子处理。根据这些原则,按照产物选择性大小进行排序,取选择性较高的产物作为动力学模型考虑的组分。将色谱分析数据归并处理得到33种主要产物。表1列出了CAT-LC和CAT-SC 2种催化剂催化下,在反应温度550℃、停留时间7s时,MCHA催化裂化反应主要产物的摩尔流率。
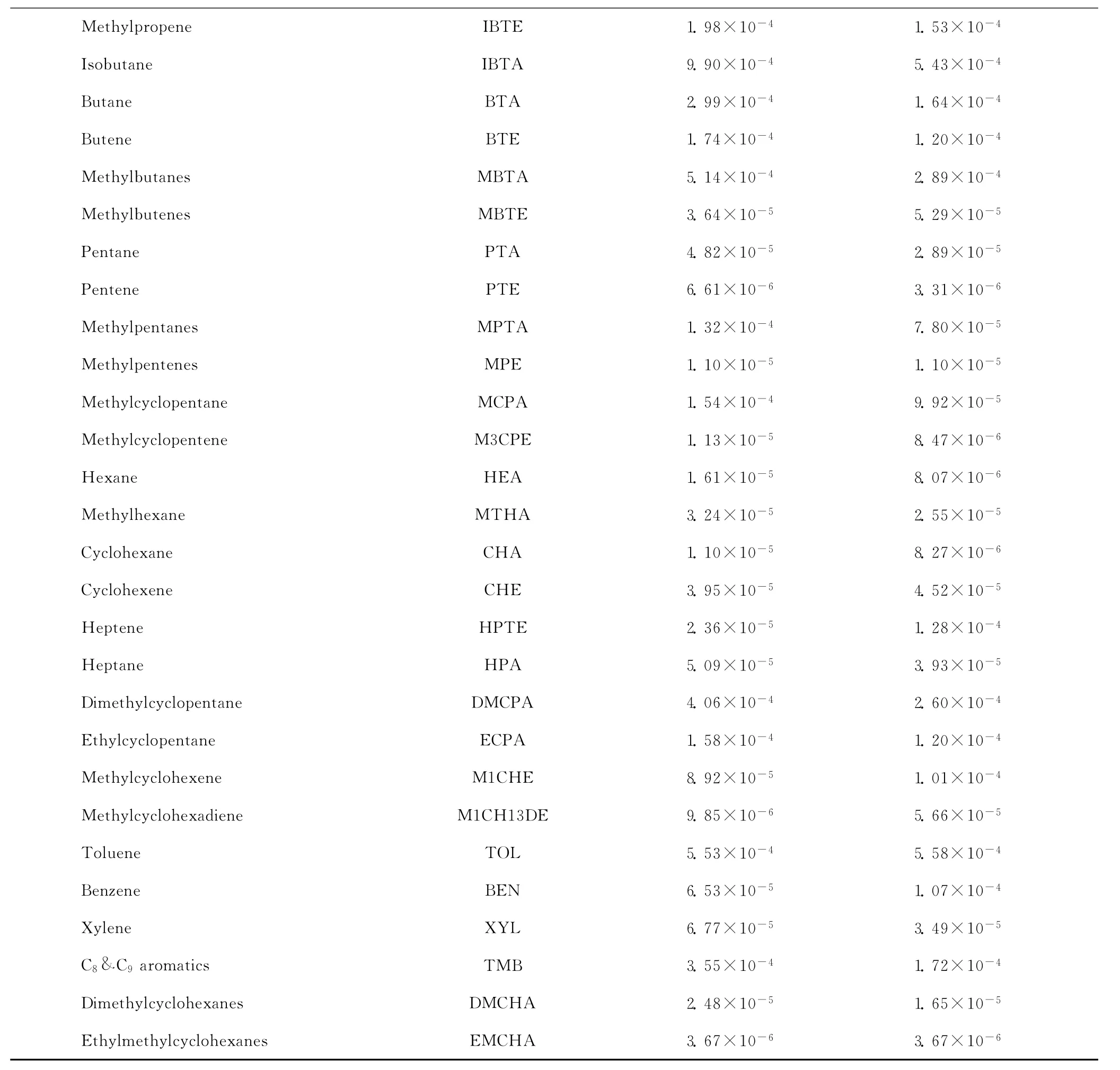
续表1
3.2 反应机理分析
MCHA催化裂化反应主要是由氢转移、异构化、质子化开环裂化、β-断裂、脱烷基和烷基转移6个主要反应类型所组成的复杂的串联和平行反应[16]。MCHA分子中包含5个仲碳原子、1个伯碳原子和1个叔碳原子,可以发生开环反应,生成烷烃和烯烃;以H+进攻叔碳原子形成叔正碳离子为主,进一步发生β-断裂和开环反应等。在酸性催化剂催化下,环烷烃的六元碳环经过质子化环丙烷异构化,发生缩环反应,生成带有1个烷基取代的五元环烷烃。另外,六元环具有较强的脱氢趋势,可形成苯、甲苯等芳烃分子,芳烃进一步脱氢缩合形成焦炭[17-18]。
3.3 反应规则的制定
反应规则不仅是一个结构能否发生某一反应或者该结构发生各种反应顺序的判据,也是判断生成产物分子所有反应的依据。它是对可能发生反应的概括和归纳,不同的工艺过程需要针对性的制定不同反应规则。
MCHA作为典型的单环环烷烃,根据催化裂化反应化学知识,针对其催化裂化反应制定了6大类共计20条反应规则。如,MCHA发生裂化反应不生成甲基、乙基正碳离子,不会同时生成多个正碳离子,不生成二烯烃,取代基都为烷烃,不区分环烷环上取代基是直链还是支链;考虑正构烯烃向异构烯烃转化,正构烷烃向异构烷烃转化,六元环向五元环的缩环异构的异构化反应;只考虑饱和环的开环反应;烯烃和环烷烃可以发生氢转移反应,生成芳烃和相应的烷烃,环状结构的氢转移反应都以生成具有芳香性的稳定结构结束;环烷烃(有取代基的环烷烃)可以发生两分子之间的烷基转移和取代烷基的脱烷基反应。
3.4 动力学参数
过渡态理论结合统计热力学可以求取MCHA催化裂化反应的指前因子。笔者采取气相分子平动熵变值近似代替反应过程中活化熵与反应物熵的差值,引入一个校正因子θ,简化了求解过程,得到了MCHA催化裂化反应中的指前因子数值[13]。在计算过程中,采用了一些归并措施[19],得到的指前因子数值与Watson等[20]的结果相吻合。不同类别反应的指前因子数值相差较大。
3.5 反应网络
以催化裂化反应化学为指导,结合制定的反应规则,对MCHA催化裂化反应建立了74个基于反应路径层面分子水平化学反应组成的反应网络。为了使反应网络具有封闭性,增加了7种重要的中间体分子,即反应网络中的分子数达到了40种。表2给出了各反应族的反应数目。与机理模型相比,反应路径层面反应数目明显减少,最多的反应类型是氢转移反应,这也与实际的环烷烃催化裂化反应规律相一致。
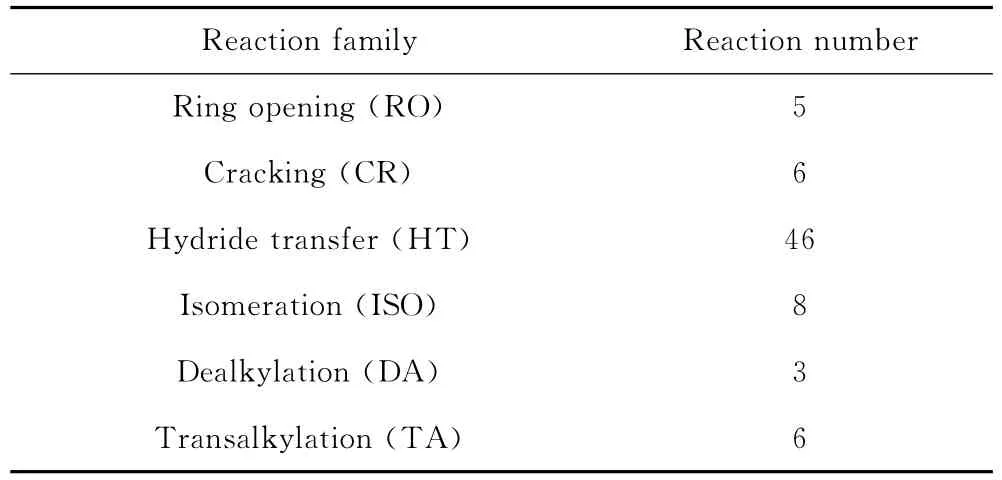
表2 MCHA催化裂化反应各反应族反应数目汇总Table 2 A summary of each reaction number of MCHA catalytic cracking
3.6 动力学模型建立与求解
当获得完整的动力学参数初值,搭建好反应网络后,运用Levenberg-Marquardt(LM)算法对不同催化剂体系下实验得到的产物分布数据进行回归计算,旨在获得一组具有普适性的动力学参数值,使建立的模型能够达到较好的预测效果。首先通过模型利用动力学参数初值以一定时间步长Δt对产品分布进行模拟计算(Simulation),随后对产物摩尔流率的预测值和实验值进行比较,若方差达到最小要求,则停止计算,否则调整动力学参数值,重复计算(Tuning)直到满足要求为止。
4 MCHA催化裂化反应动力学模型预测结果与实验结果的比较
图1为CAT-LC和CAT-SC 2种催化体系下MCHA催化裂化反应实验结果与动力学模型模拟计算的对比。从图1可以看出,主要反应产物如丙烯、异丁烷、甲苯等的摩尔流率的模拟数据与实验数据吻合良好,大部分结果分布于对角线附近,说明建立的模型具有较高的准确度。计算得到实验数据和预测数据的相关系数分别为0.92、0.82。虽然与Watson等[20]建立的辛基环己烷机理水平催化裂化反应动力学模型精度有一定的差距,他们建立的模型预测值与实验值误差在5%范围内,但是笔者建立的动力学模型不需要大量的理论计算,并且模型建立过程较为简单,扩展性较好,动力学参数可用于其他催化裂化反应体系。MCHA催化裂化反应过程中发生的化学反应成百上千,笔者仅用40种石油分子建立的74个化学反应来表示MCHA催化裂化反应过程。
图1中有个别数据相差较大,可能是由于在反应过程中忽略了一部分的化学反应,给模拟精度带来了一定影响。以MCHA开环裂化生成烯烃的反应过程为例进行详细分析,该反应过程示于图2。从图2可以看出,当MCHA生成1个正碳离子后,一部分发生环烷环缩环反应,一部分则发生开环反应。反应生成的烷烃正碳离子以及烯烃型的正碳离子发生了质子化、去质子化、异构化、甲基迁移、β-断裂等一系列反应,反应过程十分复杂,并且中间发生的化学反应大部分都是可逆反应,增加了反应的复杂程度,也给分子水平动力学模型建立带来了一定的误差。
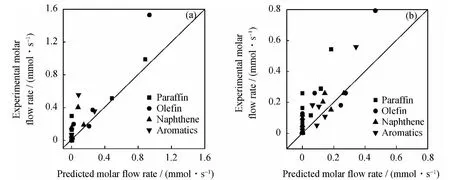
图1 CAT-LC和CAT-SC催化剂作用下MCHA催化裂化反应主要产物摩尔流率的预测值与实验值对比Fig.1 Comparison between predicted and experimental molar flow rates of main products from MCHA catalytic cracking over CAT-LC and CAT-SC catalysts
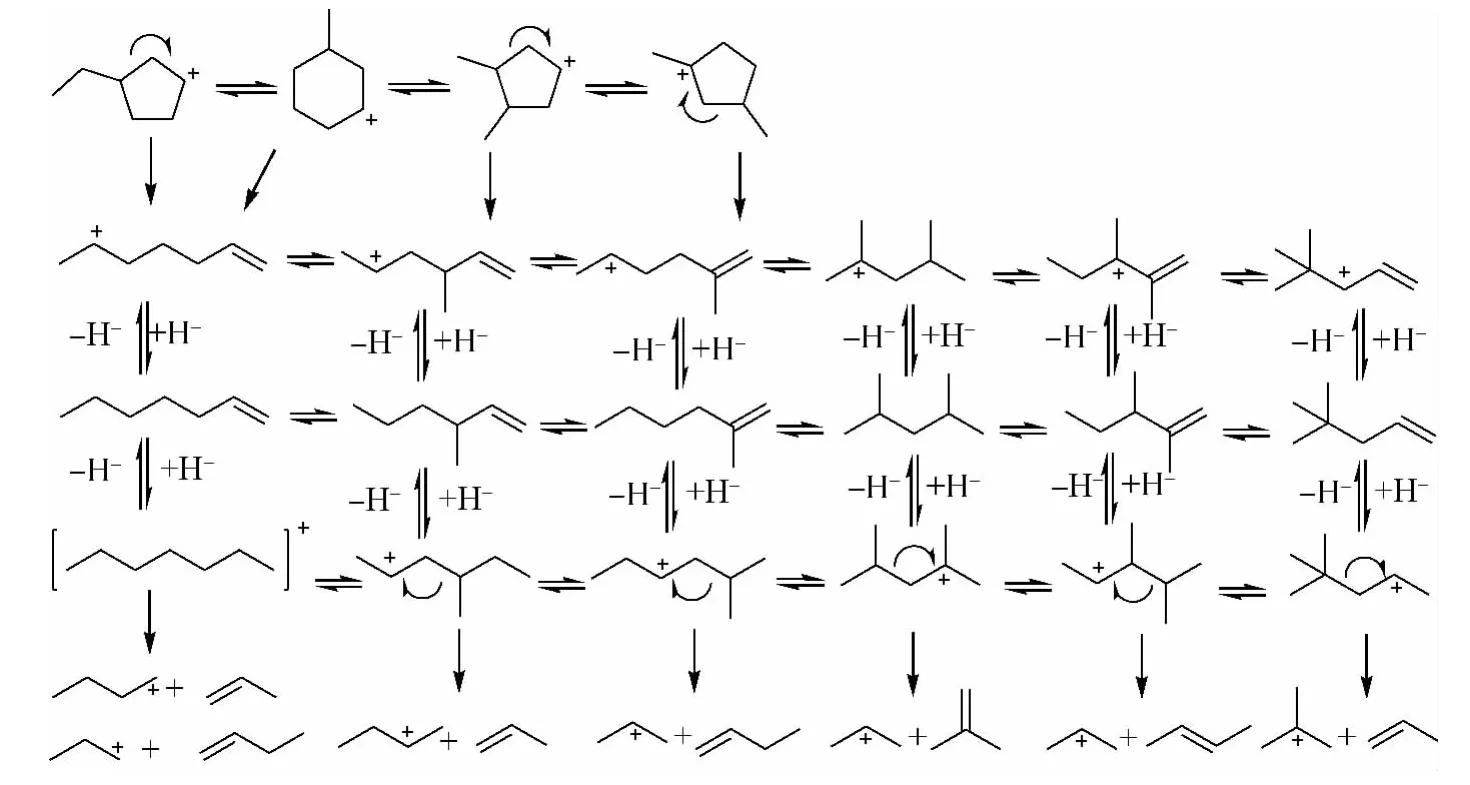
图2 MCHA开环裂化生成烯烃的反应过程Fig.2 Reaction pathways of olefin formation from MCHA ring opening
表3是回归计算得到的12类MCHA催化裂化反应动力学参数值。将从优化得到的动力学参数与先前工作中得到的初值[13]进行对比,发现两者之间数量级无大的差别,仅个别数据的差别较大(如脱烷基反应),说明其初值具有较好的准确性。对比2种催化剂E0数据可以看出,CAT-LC的催化裂化反应活性要高于CAT-SC,β裂化、异构化等反应活化能差值不大,而对于氢转移反应,2种催化剂差值较大。活化能E0值可以反映催化剂的不同[12,21]。对氢转移反应而言,CAT-LC的E0值明显低于CAT-SC,说明CAT-LC更有利于氢转移反应的发生,这主要是因为CAT-LC催化剂有较大的孔径,有利于反应物、产物分子的扩散,提高双分子氢转移反应速率[12,22],表1中产物芳烃含量较多也证明了这一点。另外,经过乙基环己烷、辛基环己烷等单环环烷烃的催化裂化实验数据验证,回归得到的12类动力学参数值,也可以用于其动力学模型的建立,这也为进一步建立单环环烷烃催化裂化反应分子水平动力学模型提供数据支持。
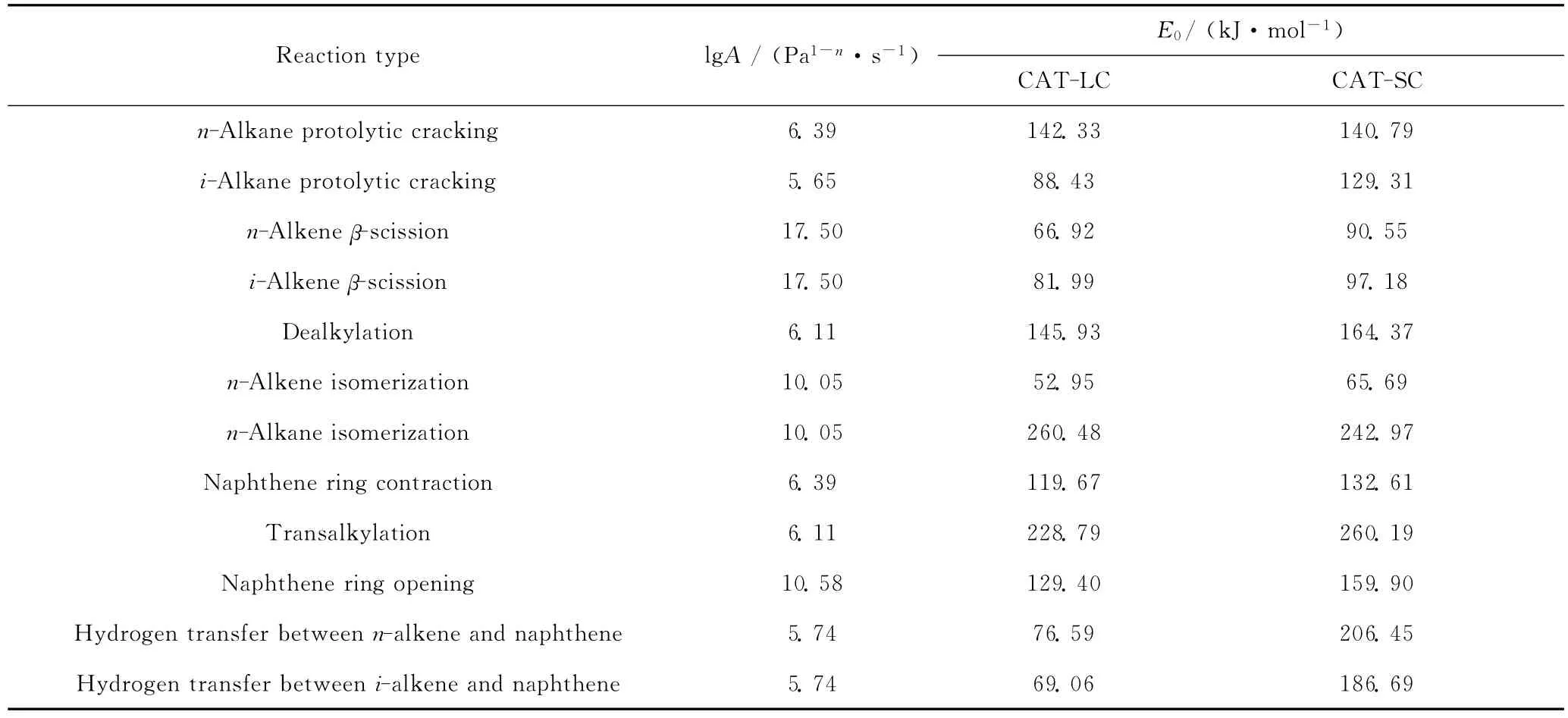
表3 回归得到的MCHA催化裂化反应动力学参数Table 3 Optimized kinetic parameters for MCHA catalytic cracking
5 结 论
采用MCHA作为单环环烷烃模型化合物和2种孔径尺寸不同的催化剂进行催化裂化反应,对产物进行了分析,选取了33种主要产物用于建立分子水平的催化裂化反应动力学模型。根据实验结果,简要分析了MCHA催化裂化6大反应类型,通过反应原料和产物的分子简化命名管理、制定20条反应规则、建立由74个化学反应组成的反应网络、获取动力学参数以及模型建立与求解等步骤,建立了MCHA催化裂化基于反应路径层面的分子水平动力学模型。建立的分子水平动力学模型对主要的反应产物摩尔流率的预测值与实验值吻合良好,相关系数分别为0.92、0.82。回归得到了12类甲基环己烷催化裂化反应动力学参数值,可以用于其他单环环烷烃模型化合物动力学模型的建立。
符号说明:
A——指前因子,Pa1-n·s-1;
Ai——i类反应的指前因子,Pa1-n·s-1;
C——组分摩尔浓度,mol/g;
Cbulk,xi——i物质在x类反应中的摩尔浓度,mol/g;
Cbulk,xj——j物质在x类反应中的摩尔浓度,mol/g;
Ccations,xi——i物质在x类反应中的离子摩尔浓度,mol/g;
Ccations,xj——j物质在x类反应中的离子摩尔浓度,mol/g;
E0——活化能,J/mol;
E0,i——i类反应的活化能,J/mol;
Ea——反应活化能,J/mol;
Ea,x——x类反应活化能,J/mol;
EA,forwardrxn——正向反应活化能,J/mol;
EA,reverserxn——逆向反应活化能,J/mol;
h——Planck常数,6.62×10-34(J·s);
Hproducts——产物的摩尔生成焓,J/mol;
Hreactants——反应物的摩尔生成焓,J/mol;
k——反应速率常数,速率系数的单位取决于反应的总级数;
kB——Boltzmann常数,1.38×10-23(J/K);
kij——i类反应中j的反应速率,速率系数的单位取决于反应的总级数;
kcatalytic,x——x类反应的速率常数;
kcatalytic,y——y类反应的速率常数;
n——反应分子数;
R——理想气体常数,8.314J/(K·mol);
t——反应时间,s;
T——反应温度,K;
Δn——过渡态与反应物之间的分子数变化,单分子反应为0,双分子反应为-1;
ΔHrxn,j——j反应的反应焓变,J/mol;
ΔHrxn——反应焓变,J/mol;
ΔS0*——气相活化熵差,J/(mol·K);
α——反应热与活化能之间的关联系数,量纲为1;
αi——i类反应活化能与反应热之间的关联系数,量纲为1。
[1]STANISLSUS A,MARAFI A,RAN M S.Recent advances in the science and technology of ultra low sulfur diesel(ULSD)production[J].Catal Today,2010,153(1-2):1-68.
[2]张结喜,齐艳华,邱建章.催化裂化关联模型的研究[J].计算机与应用化学,2007,24(11):1519-1522.(ZHANG Jiexi,QI Yanhua,QIU Jianzhang.Study on FCC correlation models[J].Comput Appl Chem,2007,24(11):1519-1522.)
[3]戴鑑,杨光福,王刚,等.催化裂化汽油改质降烯烃并多产丙烯的反应动力学模型研究[J].燃料化学学报,2008,36(4):432-437.(DAI Jian,YANG Guangfu,WANG Gang,et al.Kinetic model of FCC naphtha reformulation for reducing olefin and enhancing propylene production[J].J Fuel Chem Technol,2008,36(4):432-437.)
[4]许友好,龚剑波,张久顺,等.多产异构烷烃的催化裂化工艺两个反应区概念实验研究[J].石油学报(石油加工 ),2004,20 (4):1-5. (XU Youhao, GONG Jianhong,ZHANG Jiushun,et al.Experimental study on“two reaction zone”conception connected with MIP process[J].Acta Petrolei Sinica(Petroleum Processing Section),2004,20(4):1-5.)
[5]王建平,许先焜,翁惠新,等.加氢渣油催化裂化14集总动力学模型的建立[J].化工学报,2007,58(1):86-94.(WANG Jianping,XU Xiankun,WENG Huixin,et al.Establishment of 14lumps model for fluid catalytic cracking of hydrotreated residuum[J].J Chem Ind Eng,2007,58(1):86-94.)
[6]SOTELO-BOYÁS R,FROMENT G F.Fundamental kinetic modeling of catalytic reforming[J].Ind Eng Chem Res,2009,48(3):1107-1119.
[7]JAFFE S B,FREUND H,OLMSTEAD W N.Extension of structure-oriented lumping to vacuum residua[J].Ind Eng Chem Res,2005,44(26):9840-9852.
[8]WEI W,BENNETT C A, TANAKA R,et al.Computer aided kinetic modeling with KMT and KME[J].Fuel Process Technol,2008,89(4):350-363.
[9]WATSON B A,KLEIN M T, HARDING R H.Mechanistic modeling ofn-hexadecane cracking on rare earth Y[J].Energ Fuels,1997,11(2):354-363.
[10]WATSON B A,KLEIN M T, HARDING R H.Mechanistic modeling ofn-heptane cracking on HZSM-5[J].Ind Eng Chem Res,1996,35(5):1506-1516.
[11]WATSON B A,KLEIN M T, HARDING R H.Mechanistic modeling of a 1-phenyloctane/n-hexadecane mixture on rare earth Y zeolite[J].Ind Eng Chem Res,1997,36(8):2954-2963.
[12]AL-SABAWI M N.Heterogeneous kinetic modeling of the catalytic conversion of cycloparaffins[D].Ontario:The University of Western Ontario,2009.
[13]张旭,郭锦标,周祥,等.单环环烷烃催化裂化动力学模型的建立——指前因子的计算[J].石油学报(石油加工),2013,29 (2):283-288.(ZHANG Xu,GUO Jinbiao,ZHOU Xiang,et al.Kinetic modeling of catalytic cracking of monocyclic cycloparaffins——Calculation of preexponential factors[J].Acta Petrolei Sinica (Petroleum Processing Section),2013,29(2):283-288.)
[14]沈本贤,田立达,刘纪昌.基于结构导向集总的石脑油蒸汽裂解过程分子尺度动力学模型[J].石油学报(石油加工),2010,(S1):218-225.(SHEN Benxian,TIAN Lida,LIU Jichang.A molecular kinetic model for naphtha steam cracking based on structure oriented lumping[J].Acta Petrolei Sinica(Petroleum Processing Section),2010,(S1):218-225.)
[15]杜梅西克,拉德,阿帕里西奥,等.多相催化微观动力学(沈俭一译)[M].北京:国防工业出版社,1998:25-40.
[16]张旭,周祥,郭锦标,等.甲基环己烷催化裂化的研究进展[J].石油化工,2013,42(1):104-110.(ZHANG Xu,ZHOU Xiang,GUO Jinbiao,et al.Advances in the development of methylcyclohexane catalytic cracking[J].Petrochem Technol,2013,42(1):104-110.)
[17]于珊,张久顺,魏晓丽.环烷烃催化裂解生成乙烯和丙烯反应探析[J].石油学报(石油加工),2013,29(3):475-481.(YU Shan,ZHANG Jiushun,WEI Xiaoli.Exploration and analysis on ethylene and propylene formation in naphthene catalytic cracking[J].Acta Petrolei Sinica(Petroleum Processing Section),2013,29(3):475-481.)
[18]YU S,ZHANG J S,WEI X L.Research on ethylene and propylene formation during catalytic pyrolysis of methylcyclohexane[J].China Pet Process Pe,2012,14(4):73-79.
[19]LEE J H,KANG S,KIM Y,et al.New approach for kinetic modeling of catalytic cracking of paraffinic naphtha[J].Ind Eng Chem Res,2011,50(8):4264-4279.
[20]WATSON B A,KLEIN M T, HARDING R H.Catalytic cracking of alkylcyclohexanes:Modeling the reaction pathways and mechanisms[J].Int J Chem Kinet,1997,29(7):545-560.
[21]Al-SABAWI M,DE LASA H.Kinetic modeling of catalytic conversion of methylcyclohexane over USY zeolites:Adsorption and reaction phenomena[J].AIChE J,2009,55(6):1538-1558.
[22]唐津莲,许友好,汪燮卿,等.十氢萘在分子筛催化剂上的开环反应研究[J].燃料化学学报,2012,40(12):1422-1428. (TANG Jinlian, XU Youhao, WANG Xieqing,et al.Opening of naphthenic ring in decalin cracking over zeolite catalysts[J].J Fuel Chem Technol,2012,40(12):1422-1428.)
