樟树5种化学类型叶片转录组分析
2014-11-17江香梅伍艳芳肖复明熊振宇徐海宁
江香梅,伍艳芳,肖复明,熊振宇,徐海宁
江西省林业科学院,国家林业局樟树工程技术研究中心,南昌 330032
樟树(Cinnamomum camphora(L.) Presl)是我国特有国家Ⅱ级保护树种,是集材用、药用、香料、油用、化工、观赏、生态环境和生态文化建设等于一体的多用途树种,可作为樟科的代表种,具有极重要的开发利用价值[1]。樟树的化学成分复杂多样,其根、茎、叶、花、果中均富含精油,从其精油中已鉴定出 150余种化合物,且新化合物还在不断被鉴定出来。我国是世界上生产樟油最多的国家,其产量占世界的 80%,产品质量享誉全球。樟树按叶精油主要化学成分种类及含量的不同,可分为芳樟(精油中主成分为芳樟醇,下同)、脑樟(樟脑)、油樟(1,8-桉叶油素)、异樟(异-橙花叔醇)和龙脑樟(右旋龙脑)5种主要化学类型[2]。迄今为止,对樟树精油化学成分分析与利用的研究较多[3~5],而对化学成分代谢途径的研究则较少[6,7]。目前,以樟树为代表的樟科植物尚未完成全基因组测序,EST文库尚未建立,GenBank上樟树EST序列也较少,相关基因的功能注释亦不完善,给樟树萜类化合物次生代谢途径的研究带来极大困难,亟需更多的基因组或转录组信息来解决这一问题。
新一代测序技术(Next-generation sequencing technologies,NGSTs)对分子生物学的发展起到了巨大的推动作用。其中用于转录组测序和数字基因表达谱(Digital gene expression profiling,DGE)研究的RNA-seq技术不仅能够广泛应用于有参考基因组序列的物种研究,如水稻(Oryza sativa)[8]、葡萄(Vitis vinifera)[9]、黄瓜(Cucumis sativus)[10]等,也能应用于无参考基因组序列的物种,如青蒿(Artemisia annua)[11]、人参(Panax ginseng)[12]、红豆杉(Taxus mairei)[13]、罗汉果(Siraitia grosvenorii)[14]等,应用较为广泛[15]。基于转录组测序得到的基因功能注释、蛋白编码区注释等大量的信息,可以从基因的表达水平[9,13]、SNP鉴定[11,16]、SSR分子标记筛选[17,18]、候选基因的挖掘[19,20]、融合转录本的表达[8,21]、可变剪接[8,22]等方面展开相关研究,并建立相关转录组数据库[17],为进一步研究提供重要基础。本研究采用 Illumina HiSeq™ 2000新一代高通量测序技术对樟树5种化学类型叶片转录组进行测序,对测序得到的大量Unigene进行GO、COG和KEGG分类统计,给出功能注释和Pathway注释,并预测Unigene蛋白CDS。这些注释信息的完成将为樟树精油主要成分合成、功能基因及相关候选基因的发掘提供基础数据,同时也为进一步克隆功能基因全长、研究基因功能奠定了基础,对推动我国樟科植物分子生物学研究将起到极大促进作用。
1 材料和方法
1.1 材 料
试验材料采自江西省林业科学院内年龄约 30年的樟树成年植株。于4月底统一采集脑樟、芳樟、油樟、异樟、龙脑樟 5种化学类型幼嫩叶片,液氮速冻后-70℃低温保存,用于提取RNA,测序。
1.2 方法
1.2.1 RNA提取、纯化和文库构建
采用通用植物总RNA提取试剂盒RNeasy Plant Mini Kit提取樟树5种化学类型叶片总RNA,琼脂糖凝胶电泳检测RNA完整性,Agilent 2100 Bioanalyzer检测总 RNA 浓度。用 Oligotex® mRNA kit(TaKaRa)分离纯化 mRNA。得到的 mRNA采用fragmentation buffer打断成小片段,经过PCR扩增,建立小片段测序文库。文库测序采用 Illumina Hi-Seq™ 2000完成。
1.2.2 序列拼接
测序后得到的Raw reads,去除含有带接头的、重复的(N>5%)、测序质量很低的reads(质量值Q≤10的碱基数占整个 reads的 20%以上),获得 Clean reads。采用短 reads组装软件 Trinity[23]做转录组从头组装。先将具有一定长度overlap的reads连成更长的片段,得到Contig组装片段,再将reads比对回Contig,通过 paired-end reads来确定来自同一转录本的不同Contig以及这些Contig之间的距离,将这些 Contig连在一起,得到两端不能再延长的序列,即为Unigene。
1.2.3 功能注释和CDS预测
功能注释信息给出Unigene的蛋白功能注释、COG功能注释。先通过BlastX将Unigene序列比对到蛋白数据库 Nr(NCBI非冗余蛋白库)、SwissProt(去冗余的蛋白数据库)、KEGG(系统分析基因产物在细胞中的代谢途径以及这些基因产物功能的数据库)和 COG(对基因产物进行直系同源分类的数据库)(E值<1e-5),再通过 BlastN将Unigene比对到核酸数据库 Nt(NCBI非冗余核酸数据库)(E值<1e-5),得到跟给定 Unigene具有最高序列相似性的蛋白,从而得到该 Unigene的蛋白功能注释信息。采用 Blast2GO软件[24],根据Nr注释信息得到 GO注释信息; 采用 WEGO软件[25]对 All-Unigene(按分子功能、细胞组分、生物学过程)进行GO功能分类统计,从宏观上认识樟树的基因功能分布特征。具体测序数据分析参照林萍等[26]的转录组注释分类方法。将 Unigene序列按 Nr、SwissProt、KEGG和 COG的优先级顺序做BlastX比对(E值<1e-5)。取比对结果中rank最高的蛋白确定该 Unigene的编码区序列,根据标准密码子表将编码区序列翻译成氨基酸序列,从而得到该Unigene编码区的核酸序列(序列方向5′→3′)和氨基酸序列。与 4个数据库皆比对不上的Unigene用ESTscan[27]软件预测其编码区,得到其编码区的核酸序列(序列方向5'→3')和氨基酸序列。
2 结果与分析
2.1 樟树5种化学类型叶片转录组测序产量
樟树 5种化学类型叶片转录组测序后得到的Raw reads及去除杂质之后的Clean reads结果列于表1。由表1可以看出,质量参数Q20最低为95.08%(异樟),最高达到 98.51%(脑樟); 过滤后不确定的碱基比例N值≤0.01%; 5种化学类型的GC含量在46.55%~49.29%之间。
2.2 樟树5种化学类型叶片转录组组装质量
2.2.1 数据统计
采用短reads组装软件Trinity从头组装的樟树5种化学类型序列重叠群Contig和Unigene数目列于表 2。由表 2可知,经过拼接最终获得长度在 100~2 000 bp之间的Unigene 156 278个,总长87.09 Mb,序列平均长度584 bp,N50为1 023 bp。
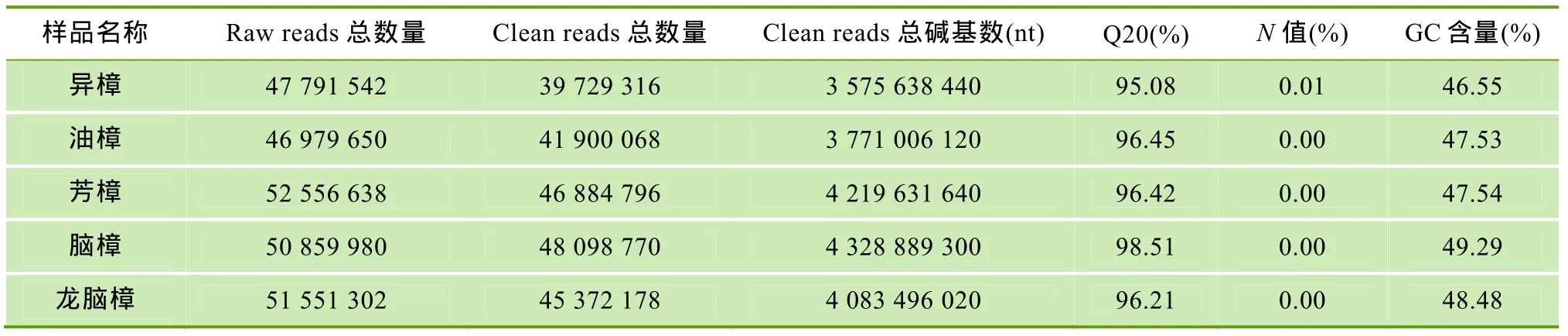
表1 测序产量统计表
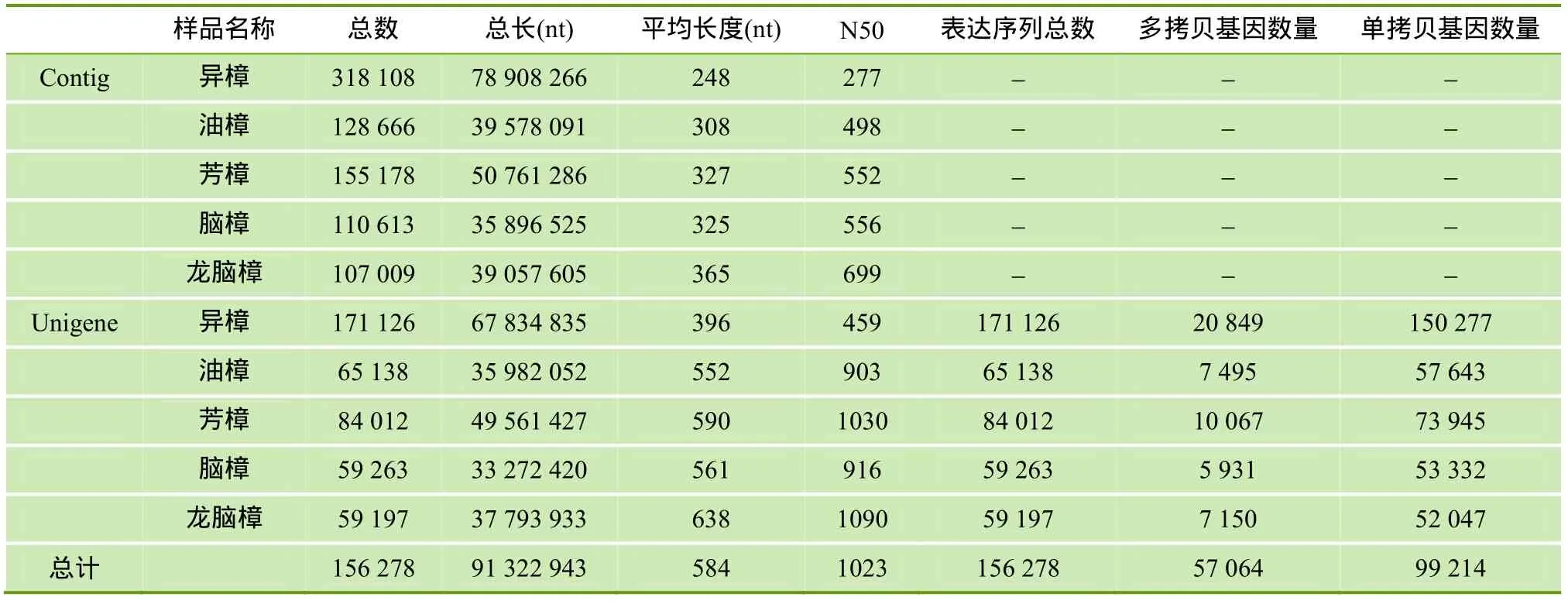
表2 组装质量统计表
2.2.2 Contig和Unigene的长度分布
Contig和Unigene的长度分布分别见图1和图2。樟树All-Unigene长度在100~500 bp的有114 115个,占73.02%; 500~1 000的有18 316个,占11.72%;1 000~1 500的有9 144个,占5.85%; 1 500~2 000的有6 365个,占4.07%; ≥2 000的有8 338个,占5.34%。
2.3 Unigene的功能注释
2.3.1 注释结果统计
分别将 Unigene注释到 Nr、Nt、SwissProt、KEGG、COG、GO库,并分别对注释到每个库以及所有注释上的Unigene数目进行统计,结果见表3。通过Blast搜索比对,共有55 955条Unigene获得了基因注释,占All-Unigene的35.80%; 有100 323条Unigene(64.20%)未被注释。Nr数据库比对注释的信息最多,注释了55 257条Unigene,COG注释的信息最少,仅21 806条Unigene得到了注释。
2.3.2 Unigene的COG分类
为了进一步评价转录组文库的完整性和注释的有效性,对Unigene进行了COG分类(图3)。在COG 25个类别中,“一般功能基因”为最大 5的组(7 466,34.24%),其次是“转录”(4 955,22.72%)和“复制、重组和修复”(3 803,17.44%)。3个最小的组别分别为“核苷酸结构”(2,0.01%)、“细胞外结构”(11,0.05%)和“RNA 加工与修饰”(358,1.64%)。此外,参与次生代谢生物合成、运输和分解的有 1 296个Unigene,占5.94%。
2.4 Unigene的GO分类
在已经得到的 Nr注释信息基础上,采用Blast2GO获得樟树Unigene的GO分类信息,共有24 717条Unigene得到GO注释。在GO分类体系中,生物学过程、细胞组分和分子功能 3个大的类别被划分为详细的 44个小的类别,其中“细胞”(16 006,14.52%)、“细胞要素”(14 515,13.17%)和“细胞器”(11 547,10.48%)3个类群占了主要部分,随后是“催化活性”(11 074,10.05%)、“结合活性”(10 642,9.65%)和“代谢过程”(9 930,9.01%)3 个类群,而“细胞杀伤”(1,0.001%)、“律动过程”(2,0.002%)和“氮素利用”(3,0.003%)仅有非常少的基因归入,这一分类结果显示了樟树叶基因表达谱的总体情况(图4)。
2.5 KEGG pathways分析
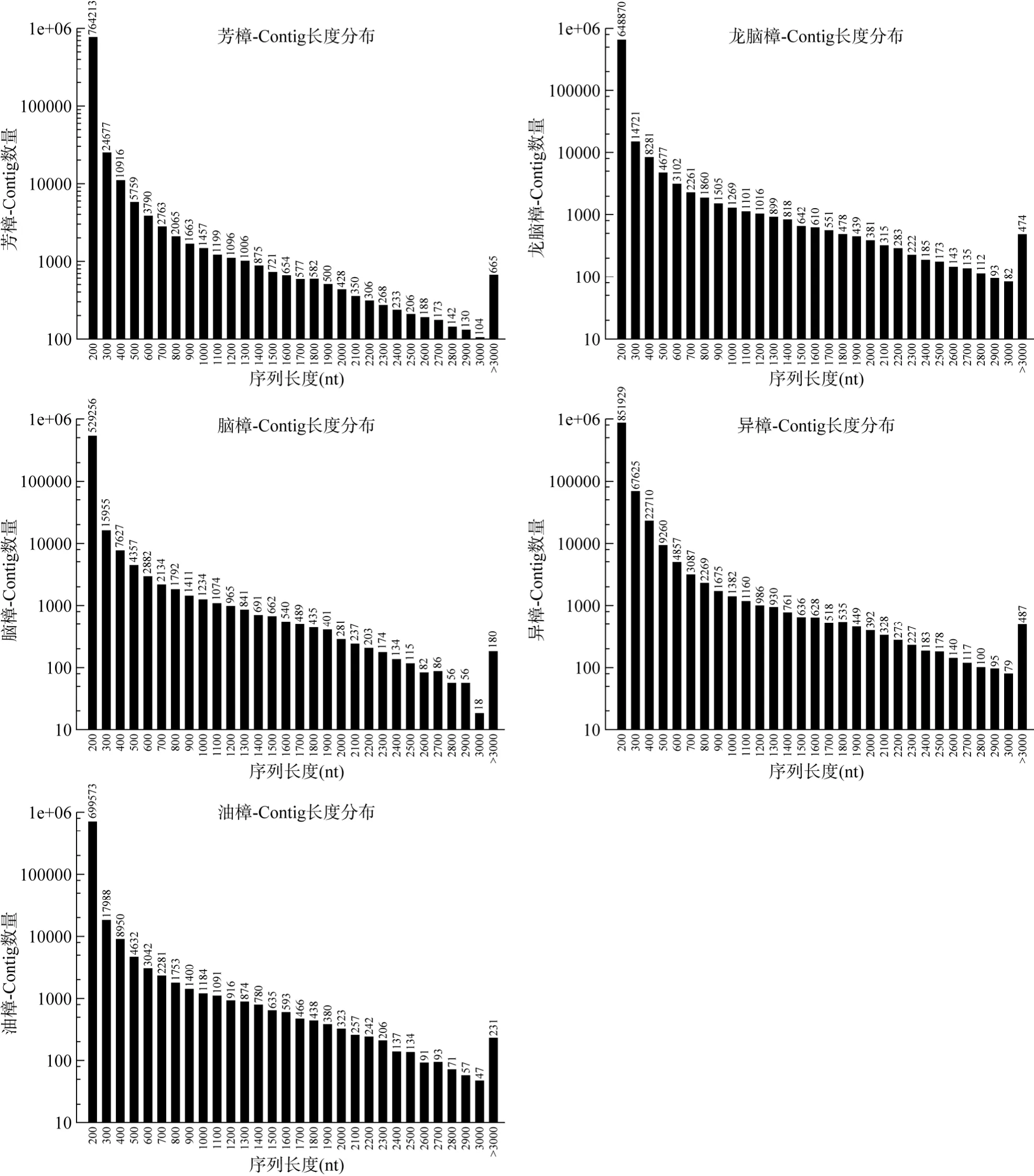
图1 Contig的长度分布统计图
利用KEGG数据库进行了功能分类和Pathway注释[28]。首先,将156 278条All-Unigene采用BlastX比对到KEGG数据库,结果有48 875条序列能够比对上(E值<1e-5),共有32 885条Unigene能够注释到217个KEGG标准Pathway,另外123 393条序列则没有相应的生物学 Pathway注释(Condition:expect≤1e-5; rank≤5),选取注释基因比例大于1%(占所有注释基因)的Pathway列于表4。由表4可知,注释到“代谢途径”中的Unigene最多,有7 808条,占23.74%; 有3 350条基因(10.19%)注释到次生代谢生物合成途径,其中,参与单萜[PATH:ko00902](66,0.2%)、二萜[PATH:ko00904](113,0.34%)、倍半萜[PATH:ko00909](34,0.1%)和萜类骨架合成[PATH:ko00900](211,0.24%)的Unigene共424个。参与不饱和脂肪酸生物合成的 Unigene有 191条,占0.58%。这些有代表性的注释为研究樟树特殊生物学进程、功能和代谢提供了重要依据。
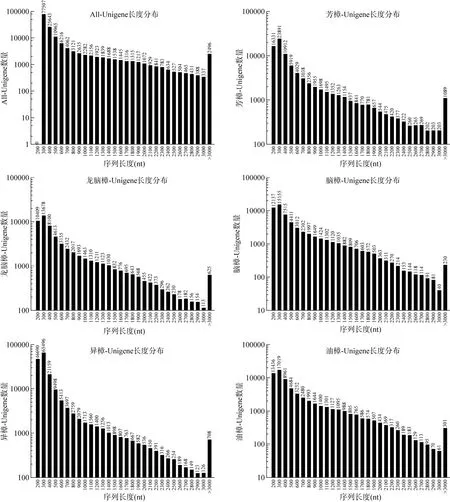
图2 Unigene的长度分布统计图
2.6 Unigene的编码蛋白框(CDS)预测
将 Unigene序列按 Nr、SwissProt、KEGG和COG数据库的优先级顺序分别做BlastX比对(E值<1e-5),确定该 Unigene的编码区序列,然后根据标准密码子表将编码区序列翻译成氨基酸序列,从而得到该Unigene编码区的核酸序列(序列方向 5′→3′)和氨基酸序列。最后,跟以上 4个数据库皆比对不上的Unigene用ESTscan软件预测其编码区,得到其编码区核酸序列(序列方向为 5′→3′)和氨基酸序列。图 5分别显示All-Unigene与数据库Blast的CDS核酸和氨基酸序列分布,ESTscan预测编码区的核酸和氨基酸序列的长度分布。
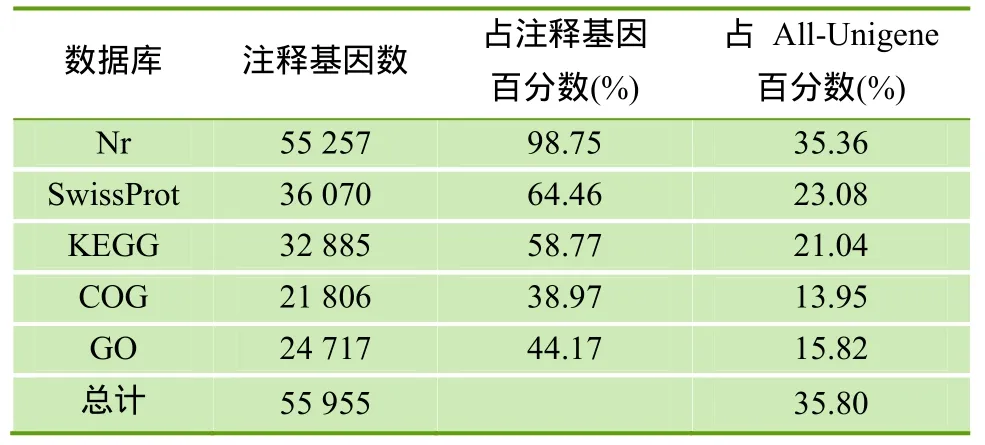
表3 Unigene注释结果统计表
2.7 樟树芳樟醇合酶基因在不同化学类型中的表达模式分析
樟树5种化学类型精油的主要成分为萜类物质,且多数为单萜和倍半萜类。在 Pathway单萜合成的代谢通路中,找到9条Unigene可能编码樟树芳樟醇合成途径的关键酶芳樟醇合酶基因,它们在 5种化学类型中的表达水平(Reads Per kb per Million reads,RPKM值)见表5。表5显示,芳樟醇合酶基因在芳樟中优势表达,而在油樟中表达水平较低。这些Unigene的注释信息将为进一步克隆功能基因的全长、研究其功能提供基础数据,也为樟树精油的代谢调控研究奠定了基础。
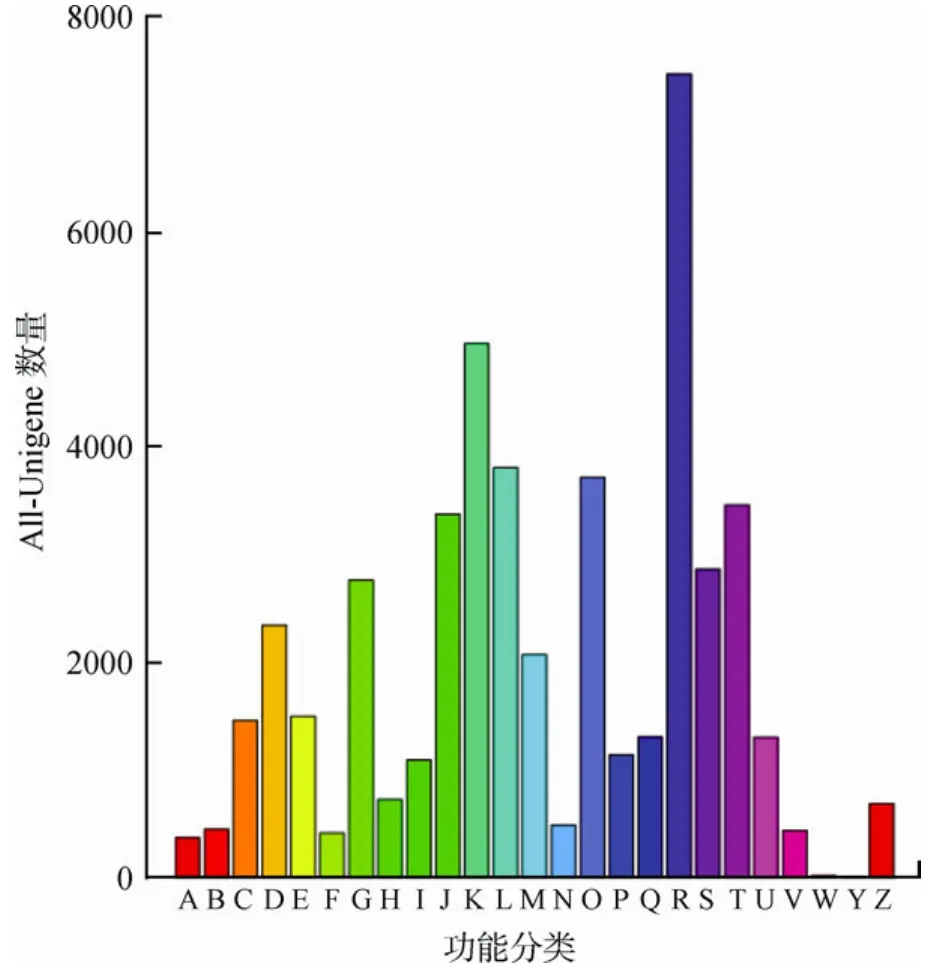
图3 Unigene的COG分类图
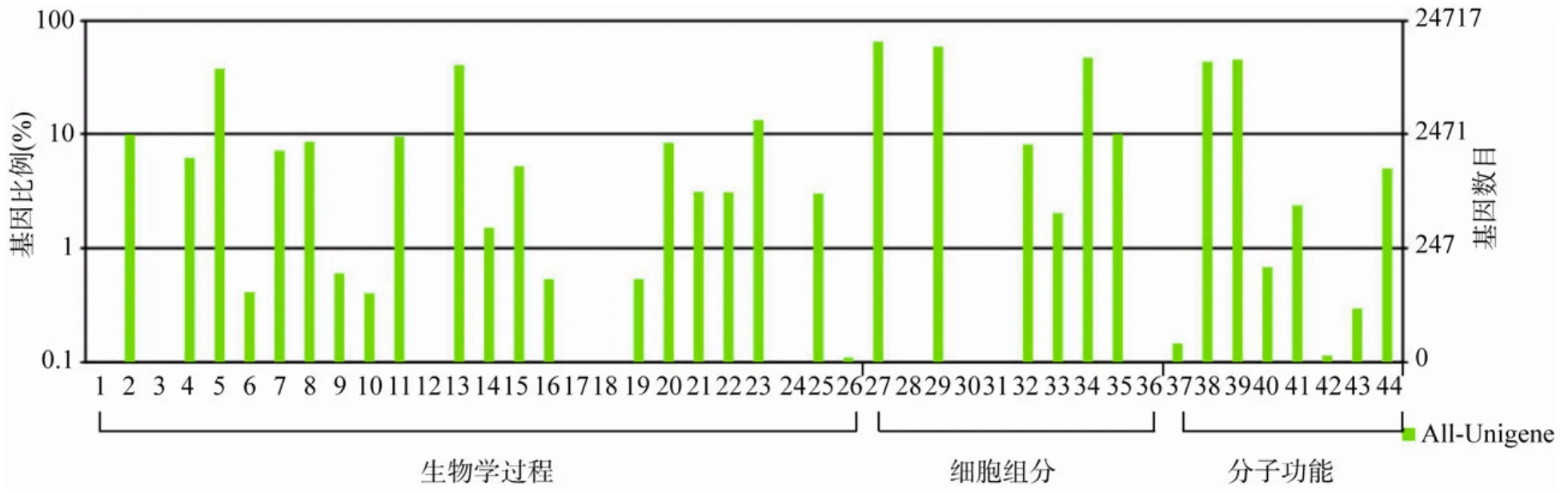
图4 Unigene的GO分类图
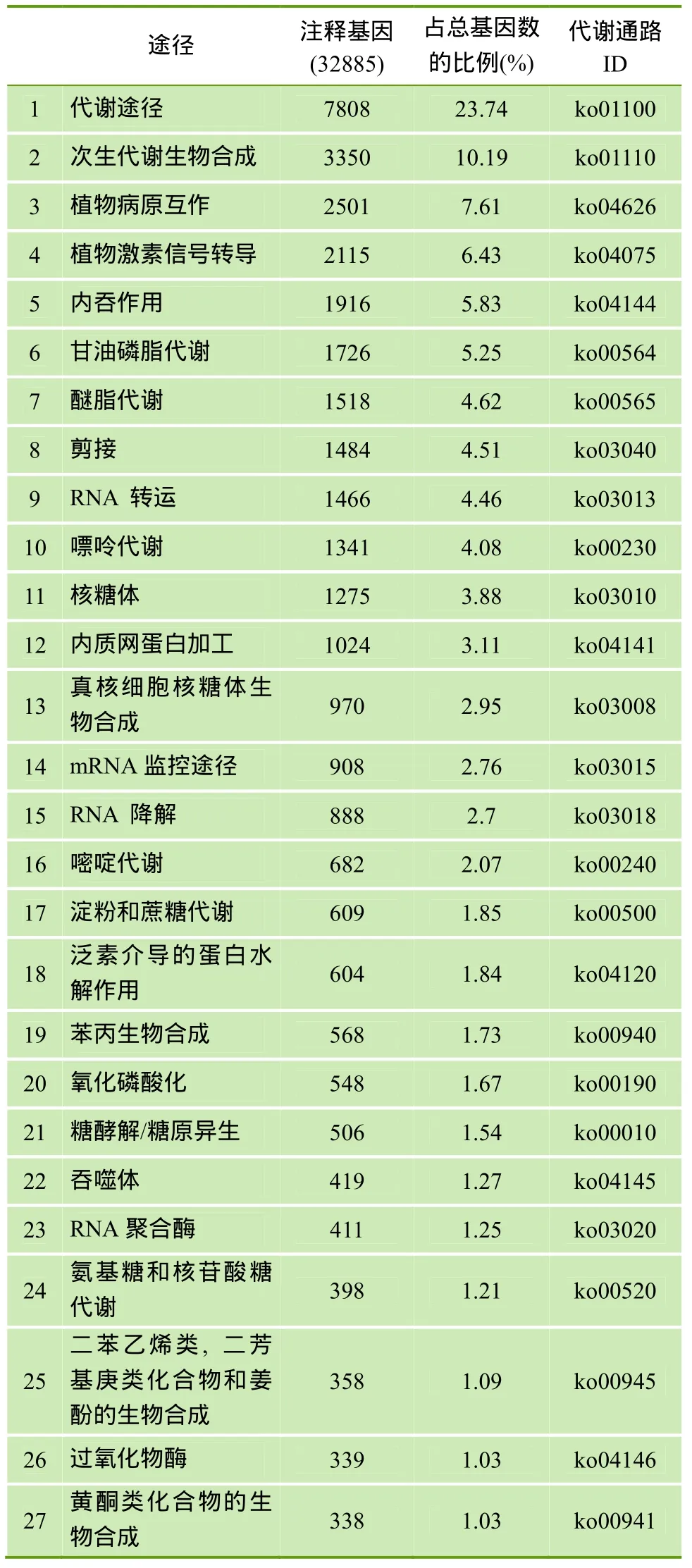
表4 KEGG pathway注释结果统计表
3 讨 论
转录组是功能基因组研究的一个重要功能指标[29]。本研究首次采用 Illumina高通量测序技术对樟树 5种化学类型叶片转录组进行测序,共拼接得到156 278条Unigene。其中,未获得鉴定的新Unigene 100 323条,占总数的64.20%,为进一步挖掘并鉴定新的功能基因提供了丰富的数据信息。本研究测序所得到的Unigene序列平均长度584 bp,N50为1 023 bp,完全满足转录组测序的要求。传统的基因表达序列标签(Expressed sequence tags,ESTs)技术被认为是一种研究转录组的有效方法,广泛应用于新基因发现、基因表达分析和蛋白质组学。基因芯片(Gene chip)技术也广泛用于大量核酸分子的检测分析,为研究不同层次多基因协同作用提供手段。与EST 技术及基因芯片技术相比,基于 Illumina HiSeqTM2000 高通量测序对转录组进行比较和分析,所需的RNA量较少,背景噪音比基因芯片低,绘制转录组遗传图谱所需的费用更低。该平台可同时用于有(无)参考基因组的转录组测序,通过短序列组装软件Trinity获得的信息量完全可满足研究需求。
木本植物由于多数为异交物种,杂合性较强,基因组相对较大且较为复杂,从而导致遗传背景研究相对滞后。而对于遗传背景不清晰的木本植物,可先采用高通量测序技术进行转录组测序,以获得的大量 Unigene信息构建遗传和物理图谱,为待测序的物种提供遗传背景信息[31]。林萍等[27]曾采用Illumina的Solexa技术对普通油茶种子4个发育时期的转录组进行测序,经组装分析获得 80 310 条Unigene,其中确定编码蛋白功能的有 21 789 条,占All-Unigene 的27.13%。本研究中,Unigene注释结果显示共有 55 955条 Unigene获得了基因注释,占 All-Unigene的 35.80%,略高于油茶转录组的注释,但是相对于草本植物来说注释结果偏低,表明现有数据库中,樟科植物基因注释信息量极为缺乏。樟树作为樟科植物的代表树种,其转录组测序及相关注释具有极为重要的意义,将为樟科其他物种提供注释信息参考。高通量测序技术可以大规模的对生物体组织样本进行测序分析,有利于建立特定时空条件下的物质代谢途径[29]。Sun等[19]对西洋参(Panax quinquefoliusL.)转录组进行测序,KEGG分析确定有 4 097条序列被定位到特定的代谢途径中,并且初步确定了从 acetyl CoA开始经过类异戊二烯途径的所有参与人参皂苷骨架合成的酶。此外,东北红豆杉(Taxus cuspidataSieb.et Zucc.)中的紫杉醇[32]、蛇足石杉(Huperzia serrata(Thunb.ex Murray)Trev.)和龙骨马尾杉(Phlegmariurus carinatus(Desv.ex Poir.) Ching.)中的石松生物碱[33]等物质也通过高通量测序进一步确定其次生代谢途径。樟树Unigene的GO分类结果显示了樟树5种化学类型叶基因表达谱的总体情况,共有24 717条Unigene得到GO注释,在 44个小类别中,归入“细胞”和“细胞器”类别的分别为 16 006(14.52%)和 11 547(10.48%)条Unigene,说明有较多的基因与细胞和细胞器中的生物代谢相关。KEGG pathways分析结果表明,共有3 350(10.19%)条基因注释到次生代谢生物合成途径,其中,参与萜类代谢的Unigene共424条。在此基础上进行芳樟醇合成途径分析,结果显示芳樟醇合酶基因在芳樟中优势表达,而在油樟中表达水平较低,这一结果与樟树 5种化学型中芳樟醇的含量高低相一致[2]。这些有代表性的注释为研究樟树次生代谢过程、脂肪酸合成途径及其他特殊生物学进程提供了重要依据。

图5 CDS的长度分布统计图
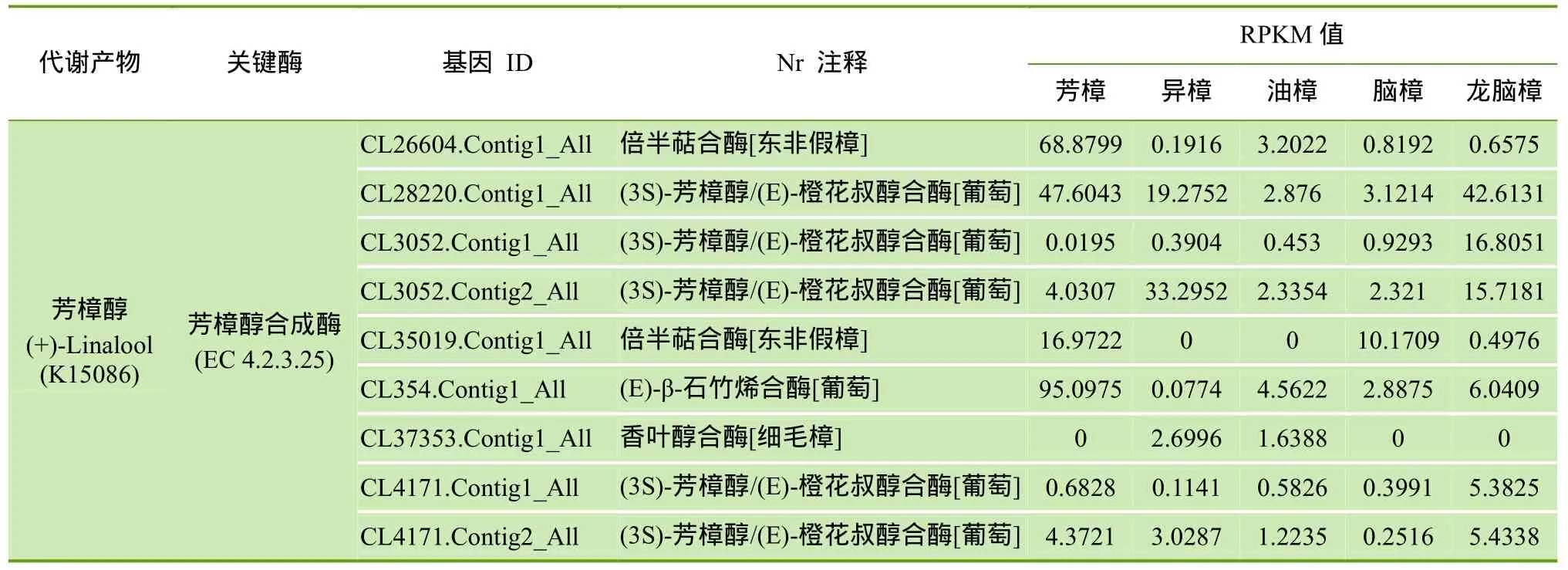
表5 编码芳樟醇合酶的Unigene注释结果统计表
[1]戴宝合.野生植物资源学.北京:农业出版社,1993.
[2]石皖阳,何伟,文光裕,郭德选,龙光远,刘银苟.樟精油成分和类型划分.植物学报,1989,31(3):209–214.
[3]彭东辉.樟树优良单株选择与组培研究[学位论文].福建农林大学,2004.
[4]Lee HJ,Hyuna EA,Yoon WJ,Kim BH,Rhee MH,Kang HK,Cho JY,Yoo ES.In vitroanti-inflammatory and anti-oxidative effects ofCinnamomum camphoraextracts.J Ethnopharmacol,2006,103(2):208–216.
[5]Liu CH,Mishra AK,Tan RX,Tang C,Yang H,Shen YF.Repellent and insecticidal activities of essential oils fromArtemisia princepsandCinnamomum camphoraand their effect on seed germination of wheat and broad bean.Bioresour Technol,2006,97(15):1969–1973.
[6]Yang T,Li J,Wang HX,Zeng Y.A geraniol-synthase gene fromCinnamomum tenuipilum.Phytochemistry,2005,66(3):285–293.
[7]陈美兰.药用植物樟化学型形成机理的基础研究[学位论文].中国中医科学院,2007.
[8]Zhang GJ,Guo GW,Hu XD,Zhang Y,Li QY,Li RQ,Zhang RH,Lu ZK,He ZQ,Fang XD,Chen L,Tian W,Tao Y,Kristiansen K,Zhang XQ,Li SG,Yang HM,Wang J.Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome.Genome Res,2010,20(5):646–654.
[9]Wu J,Zhang YL,Zhang HQ,Huang H,Folta KM,Lu J.Whole genome wide expression profiles ofVitis amurensisgrape responding to downy mildew by using Solexa sequencing technology.BMC Plant Biol,2010,10:234.
[10]Guo SG,Zheng Y,Joung JG,Liu SQ,Zhang ZH,Crasta OR,Sobral BW,Xu Y,Huang SW,Fei ZJ.Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types.BMC Genomics,2010,11:384.
[11]Graham IA,Besser K,Blumer S,Branigan CA,Czechowski T,Elias L,Guterman I,Harvey D,Isaac PG,Khan AM,Larson TR,Li Y,Pawson T,Penfield T,Rae AM,Rathbone DA,Reid S,Ross J,Swallwood MF,Segura V,Townsend T,Vyas D,Winzer T,Bowles D.The genetic map ofArtemisia annuaL.identifies loci affecting yield of the antimalarial drug artemisinin.Science,2010,327(5963):328–331.
[12]Chen S,Luo H,Li Y,Sun Y,Wu Q,Niu Y,Song J,Lv A,Zhu Y,Sun C,Steinmetz A,Qian Z.454 EST analysis detects genes putatively involved in ginsenoside biosynthesis inPanax ginseng.Plant Cell Rep,2011,30(9):1593–1601.
[13]Hao DC,Ge GB,Xiao PG,Zhang YY,Yang L.The first insight into the tissue specificTaxustranscriptome via Illumina second generation sequencing.PLoS ONE,2011,6(6):e21220.
[14]Tang Q,Ma XJ,Mo CM,Wilson IW,Song C,Zhao H,Yang YF,Fu W,Qiu DY.An efficient approach to findingSiraitia grosvenoriitriterpene biosynthetic genes by RNA-seq and digital gene expression analysis.BMC Genomics,2011,12(1):343.
[15]Wilhelm BT,Landry JR.RNA-Seq quantitative measurement of expression through massively parallel RNA Sequencing.Methods,2009,48(3):249–257.
[16]Zenoni S,Ferrarini A,Giacomelli E,Xumerle L,Fasoli M,Malerba G,Bellin D,Pezzotti M,Delledonne M.Characterization of transcriptional complexity during berry development inVitis viniferausing RNA-Seq.Plant Physiol,2010,152(4):1787–1795.
[17]Wang ZY,Fang BP,Chen JY,Zhang XJ,Luo ZX,Huang LF,Chen XL,Li YJ.De novoassembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato(Ipomoea batatas).BMC Genomics,2010,11(1):726.
[18]李滢,孙超,罗红梅,李西文,牛云云,陈士林.基于高通量测序454 GS FLX的丹参转录组学研究.药学学报,2010,45(4):524–529.
[19]Sun C,Li Y,Wu Q,Luo HM,Sun YZ,Song JY,Lui E MK,Chen SL.De novosequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis.BMC Genomics,2010,11(1):262.
[20]Logacheva MD,Kasianov AS,Vinogradov DV,Samigullin TH,Gelfand MS,Makeev VJ,Penin AA.De novosequencing and characterization of floral transcriptome in two species of buckwheat(Fagopyrum).BMC Genomics,2011,12(1):30.
[21]Francis RW,Thompson-Wicking K,Carter KW,Anderson D,Kees UR,Beesley AH.FusionFinder:A software tool to identify expressed gene fusion candidates from RNA-Seq data.PLoS ONE,2012,7(6):e39987.
[22]Li PH,Ponnala L,Gandotra N,Wang L,Si YQ,Tausta SL,Kebrom TH,Provart N,Patel R,Myers CR,Reidel EJ,Turgeon R,Liu P,Sun Q,Nelson T,Brutnell TP.The developmental dynamics of the maize leaf transcriptome.Nat Genet,2010,42(12):1060–1067.
[23]Grabherr MG,Haas BJ,Yassour M,Levin JZ,Thompson DA,Amit I,Adiconis X,Fan L,Raychowdhury R,Zeng Q,Chen Z,Mauceli E,Hacohen N,Gnirke A,Rhind N,Palma F,Birren BW,Nusbaum C,Lindblad-Toh K,Friedma N,Regev A.Full-length transcriptome assembly from RNA-Seq data without a reference genome.Nat Biotechnol,2011,29(7):644–652.
[24]Conesa A,Götz S,García-Gómez JM,Terol J,Talón M,Robles M.Blast2GO:a universal tool for annotation,visualization and analysis in functional genomics research.Bioinformatics,2005,21(18):3674–3676.
[25]Ye J,Fang L,Zheng HK,Zhang Y,Chen J,Zhang ZG,Wang J,Li ST,Li RQ,Bolund L,Wang J.WEGO:a web tool for plotting GO annotations.Nucleic Acids Res,2006,34(1):W293–297.
[26]林萍,曹永庆,姚小华,王开良,滕建华.普通油茶种子 4个发育时期的转录组分析.分子植物育种,2011,9(4):498–505.
[27]Iseli C,Jongeneel CV,Bucher P.ESTScan:a program for detecting,evaluating,and reconstructing potential coding regions in EST sequences.Proc Int Conf Intell Syst Mol Biol,1999,138–148.
[28]Kanehisa M,Goto S,Kawashima S,Okuno Y,Hattori M.The KEGG resource for deciphering the genome.Nucleic Acids Res,2004,32(1):D277–D280.
[29]梁烨,陈双燕,刘公社.新一代测序技术在植物转录组研究中的应用.遗传,2011,33(12):1317–1326.
[30]施季森,王占军,陈金慧.木本植物全基因组测序研究进展.遗传,2012,34(2):145–156.
[31]Li X,Chen GH,Zhang WY,Zhang X.Genome-wide transcriptional analysis of maize endosperm in response to ae wx double mutations.J Genet Genomics,2010,37(11):749–762.
[32]Wu Q,Sun C,Luo H,Li Y,Niu Y,Sun Y,Lu A,Chen S.Transcriptome analysis ofTaxus cuspidataneedles based on 454 pyrosequencing.Planta Med,2010,77(4):394–400.
[33]Luo HM,Li Y,Sun C,Wu Q,Song JY,Sun YZ,Steinmetz A,Chen SL.Comparison of 454-ESTs fromHuperzia serrataandPhlegmariurus carinatusreveals putative genes involved in lycopodium alkaloid biosynthesis and developmental regulation.BMC Plant Biol,2010,10(1):209.
