腓骨肌萎缩症2型(CMT2)小鼠模型的研究进展
2014-11-17于珍栾春杰顾鸣敏
于珍,栾春杰,顾鸣敏
上海交通大学医学院医学遗传学教研室,上海 200025
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)是一类较常见的遗传性周围神经疾病[1,2],全球的群体发病率约为1/2500[3]。该病主要呈常染色体显性遗传(Autosomal dominant,AD),也可呈常染色体隐性遗传(Autosomal recessive,AR)及X连锁隐性遗传(X-linked recessive,XR)[3]。CMT病人在患病初期往往表现为弓形足及杵状指,由于下肢无力而呈现跨越步态。随着疾病的进展,部分患者出现手部肌肉无力和萎缩,造成精细动作困难。末端神经功能的丧失还会导致感觉功能减退,如辨别冷热的能力下降。该病大多在青少年期起病,也有少数于中年起病[4~6]。目前,除了理疗、定制矫形支架或外科手术外,临床上还缺乏有效的治疗手段,药物治疗仅对个别亚型有一定的作用[7]。根据神经电生理和病理生理变化主要将CMT分为两种类型:脱髓鞘型和轴索型。脱髓鞘型CMT约占总数的 2/3,其中以呈AD遗传的CMT1最为多见,还包括呈AD遗传的CMT3,呈XR遗传的CMTX1和呈AR遗传的CMT4。脱髓鞘型CMT的神经电生理表现多为神经传导速度降低,神经病理表现为“洋葱头”样改变[8,9]。轴索型CMT约占总数的1/3,均为呈AD或AR遗传的 CMT2。CMT2各亚型的神经电生理表现为神经传导速度正常或轻度减慢,仅复合肌肉动作电位(cMAP)有所降低; 神经病理检查多无“洋葱头”样改变[8,9]。目前已报道51个基因突变均可导致CMT,但其中的4个基因(PMP22、GJB1、MPZ、MFN2)突变所致的CMT占总数的90%以上[7]。在CMT1中,最常见的是由外周髓鞘蛋白22基因(PMP22)重复突变所致的 CMT1A,约占所有 CMT的 36%; 其次是由髓鞘蛋白零基因(MPZ)突变所致的 CMT1B,约占CMT的8.5%。在X连锁遗传的CMT中,由间隙连接蛋白32基因(Cx32)突变所致的CMTX1约占所有CMT的10%。在CMT2中,由线粒体融合蛋白2基因(MFN2)突变所致的 CMT2A2最常见,约占所有CMT中的5%和CMT2中的10%~30%[7,10]。然而,迄今对CMT的发病机制所知甚少,特别是导致CMT2的17个基因的致病机制尚缺乏较深入的研究。现有的研究通常认为CMT2的发生与轴索信号传送障碍、线粒体膜融合异常、胞内体的运输受限、神经原纤维素的产生不足和线粒体分子伴侣的功能异常相关[11]。同时也认为轴索变性是一种长度依赖性病变,神经纤维越长,维持长距离轴浆运输所需线粒体及其所产生的能量越多,因此 CMT2首先影响四肢远端神经纤维,并逐步导致其所支配的肌肉发生萎缩[11]。
建立 CMT2动物模型,不仅有助于从动物水平阐明突变基因的生物学功能及致病机制,而且对未来的分子诊断和靶向治疗也具有重要的指导意义[12]。常用的模式生物包括大肠杆菌(Escherichia coli)、酵母(Saccharomyces cerevisiae)、线虫(Caenorhabditis elegans)、果蝇(Drosophilamelanogaster)、斑马鱼(Danio rerio)、小鼠(Mus musculus)等。但用于遗传病致病机制研究的动物首选小鼠,理由是:①小鼠是地球上最小的哺乳动物之一,易于饲养且繁殖量大; ②小鼠在生物进化上与人类相近; ③人类 99%的基因存在于小鼠,基因同源性高达 78.5%,且小鼠基因组中约93%的基因排列顺序与人类相同;④小鼠组织器官结构和细胞功能与人类相似[13]。本文主要综述了 CMT2小鼠模型的构建策略、CMT2亚型及已建立的小鼠模型,举例说明几种 CMT2小鼠模型的表型特征及分子机制,最后分析了该领域尚存在的问题。
1 CMT2小鼠模型的构建策略与方法
常用的CMT2小鼠模型包括转基因小鼠、突变基因敲除小鼠和突变基因敲入小鼠等。下面简要介绍了这些小鼠模型的构建策略和方法。
1.1 运用转基因技术构建CMT2小鼠模型
转基因小鼠(Transgenic mice)是指通过细胞工程方法(如显微注射技术)将外源目的基因注入小鼠受精卵的雄原核,再将该受精卵植入假孕小鼠体内使其生长发育,最终筛选出基因组中整合了外源基因的新生小鼠[14]。完成鉴定的小鼠称为首建鼠(Founder mice),将其进一步繁育成遗传学上稳定的小鼠品系,就可用于相关医学研究。例如,2008年中南大学湘雅医学院唐北沙课题组[15]通过位置候选克隆发现热休克蛋白 β8(HSPB8,也称HSP22)为CMT2L的致病基因,并采用显微注射技术建立了HSPB8K141N转基因小鼠模型,并对小鼠模型进行了行为学、电生理学和病理学分析,以期再现CMT2L的临床表型,用于 HSPB8 生物学功能分析和CMT2L发病机制研究。
1.2 运用基因敲除技术构建CMT2小鼠模型
基因敲除小鼠(Knock-out mice)是指利用同源重组原理,将完全失活或纠正的外源打靶基因取代内源核基因组中的目标基因的转基因小鼠[14]。目前常用的基因敲除方法有两种:(1)组成型基因剔除:即在培养的胚胎干细胞(ES细胞)中直接将目的基因敲除或破坏,导致该基因失去功能; 该法易产生胚胎致死的后果,较难获得理想的小鼠模型。(2)诱导型基因剔除:该法使目的基因的敲除发生在发育过程中人为设定的某一阶段和特定的组织细胞中。由于该法在目的基因的两侧增加了 loxP位点,同时也又增加了Cre基因,可满足定时和定点失活目的基因的要求,故能避免胚胎致死的产生,是目前首选的基因剔除技术[16]。例如,2001年Zhao等[17]报道了由该实验室构建的驱动蛋白 1B(Kif1B)基因敲除小鼠模型,以期模拟 CMT2A1的表型。基因敲除小鼠Kif1B基因组来自一个 ES细胞系 J1的λEMBL3基因库,包含P环和上游外显子2.2 kb大小的区域被一个含较少poly(A)的neo筛选标记替换,获得的嵌合体小鼠经过回交,得到的杂合子小鼠与杂合子小鼠交配获得突变纯合子小鼠模型。
1.3 运用基因敲入技术构建CMT2小鼠模型
基因敲入小鼠(Knock-in mice)是指利用同源重组原理,将外源目的基因插入到核基因组目标基因序列的内部或内源目标基因启动子之后的转基因小鼠[14]。与基因剔除技术相比,该法的优势在于可在一个目标基因的编码区若干碱基被去除的同时,另一个目标基因编码区的若干碱基被替换。采用这种方法可以研究具有完全不同结构的两个基因的产物是否具有相同的生物学功能[16]。已知髓鞘蛋白零基因(MPZ)编码一种外周髓鞘的主要结构蛋白,该基因突变会导致CMT2I/CMT2J/CMT1B等疾病。2012年Saporta等[18]报道了由该实验室构建的MpzR98C基因突变敲入小鼠模型。编码MPZR98C的突变通过同源重组被定位到Mpz等位基因上。接着,通过定点诱变和序列分析证实该突变已被引入Mpz基因第3号外显子中。克隆片段连接成含有新霉素抗性基因构造的loxP位点。构建的R98C neoLP和WT neoLP载体通过显微注射进入 TBV2胚胎干细胞。然后将阳性胚胎干细胞克隆入野生型小鼠的囊胚中获得嵌合体。通过种系繁殖及鉴定,获得遗传背景为 FVB/N或C57BL/6N的MpzR98C敲入小鼠模型。
2 CMT2亚型及其小鼠模型概览
迄今已报道的CMT2致病基因共有17个,由此产生相应的 CMT2亚型,包括本实验室近期报道的由脱氢酶 E1和转酮酶结构域 1(Dehydrogenase E1 and transketolase domain containing 1,DHTKD1)无义突变所致的CMT2Q[19]。在已经报道的CMT2亚型中,有些亚型已建立了小鼠模型,并开展了小鼠的表型分析及机制研究[20]; 有些亚型正在构建小鼠模型,为后续研究作准备。CMT2各亚型的遗传方式、OMIM 编号、疾病基因名称及定位、小鼠模型的构建方法及主要特征等见表1。
3 部分 CMT2亚型小鼠模型的表型特点及病理机制
3.1 CMT2A1小鼠模型
已知驱动蛋白 1B(KIF1B)突变会导致人类CMT2A1。该基因编码两个异构体:KIF1Bα与KIF1Bβ。Zhao等[17]发现 KIF1Bβ与 KIF1Bα的底物结合域明显不同。为了验证KIF1B基因在体内的重要功能,该实验室成功建立了Kif1B基因敲除小鼠模型,其中突变纯合子小鼠(Kif1B-/-)的出生率符合孟德尔遗传定律,但是在出生30 min内死亡。研究发现纯合子小鼠出现肺泡不能扩张,由此导致小鼠窒息和死亡。进一步研究发现新生的纯合子小鼠表现出多种神经异常,如对刺激反应不敏感,运动神经功能丧失。与野生型小鼠(Kif1B+/+)相比,纯合子小鼠大脑体积减少约 10%、脑干核心以及联合纤维的形态及发育受损、神经元数量减少(约 25%)、海马体的细胞结构及组织形态受损,突触小泡的密度明显降低。此外,背侧及腹侧骨髓网状核(MdD和MdV)神经元数量减少。杂合子小鼠(Kif1B+/-)在出生一年后才逐步表现出跨越步态,最终不能支撑自身体重,只能通过偶尔的跳跃来移动身体,表现出类似CMT2A1患者的特点。在滚轴、固定杆和握力实验中,杂合子比野生型小鼠明显减弱; 而在测定感觉的热板实验中,杂合子与野生型小鼠并没有出现显著性差异。为了进一步探究肌无力的本质,直接刺激12月龄杂合子小鼠的足底肌,以测量坐骨神经诱发的复合动作电位,结果发现杂合子小鼠的振幅有明显变化,但神经传导速度没有明显改变。另外,远端肌肉的组织病理学分析也未见明显差异。在杂合子小鼠中还观察到脑干中心及结合处的轴索发育受损并且脊柱中轴索发育受损,这一现象被认为是周围神经病变严重的特征。此外,Kif1B-/-神经元的低存活率可以通过 KIF1Bβ 的 cDNA 的表达来补救,而不是通过 KIF1Bα,表明在神经元中 KIF1Bβ表现出了自主行为,这与CMT2A1作为轴索病变而非髓鞘病变的临床意义保持一致。课题组又分析了CMT2A1患者中KIF1B的序列,发现其ATP结合结构域存在Q98L的错义突变。综上所述,KIF1Bβ蛋白的改变降低了ATPase的活性和功能,并且由该基因产生的马达蛋白含量减半,不能维持轴索所需的动力,导致神经末梢功能障碍,最后出现 CMT2A1的表型。
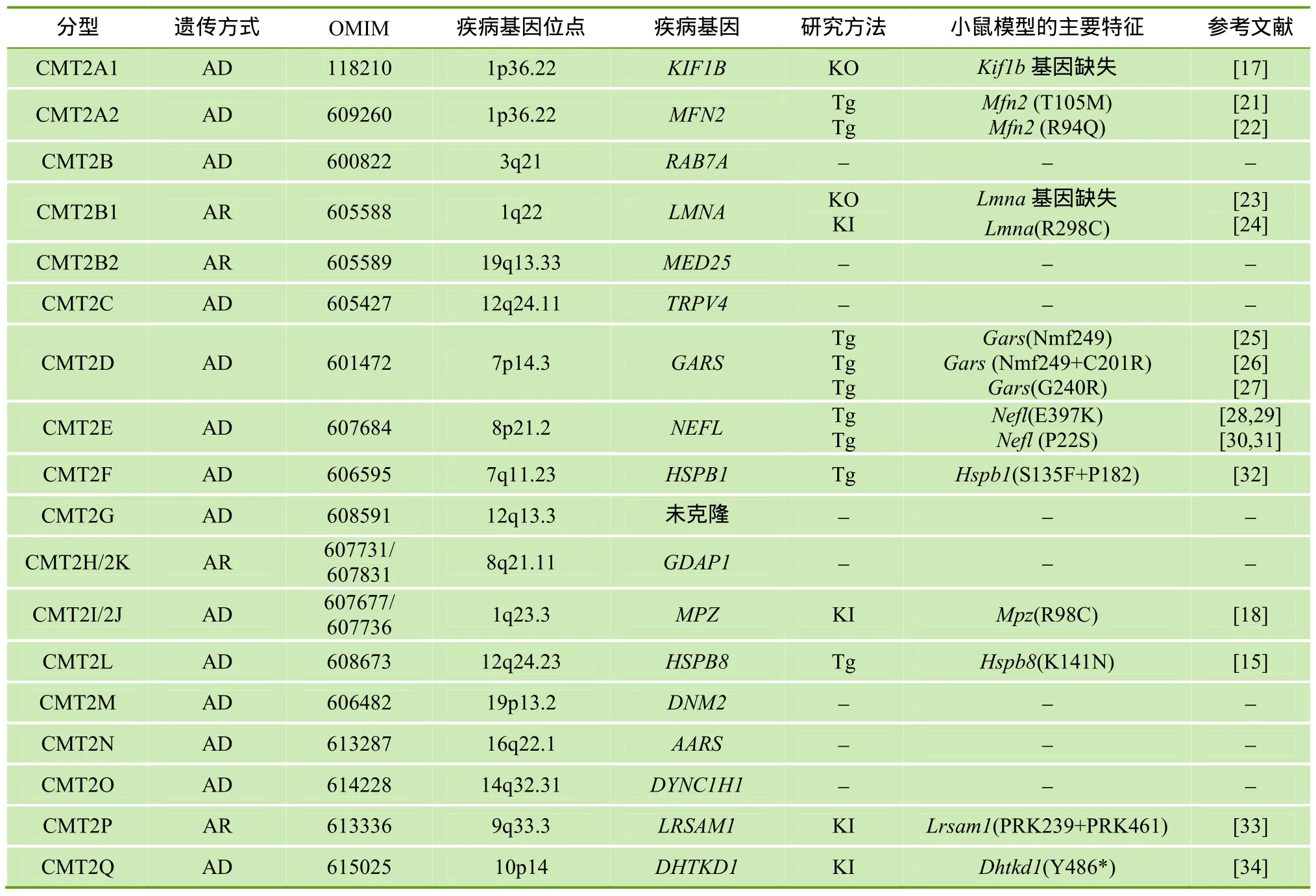
表1 CMT2亚型及其小鼠模型一览表
3.2 CMT2A2小鼠模型
1997年,Saito等[35]报道了一个日本家系患有CMT2A2。2004年,Zuchner等[36]确认了线粒体融合蛋白2基因(MFN2)为CMT2A2的致病基因。MFN2是线粒体融合中必须的GTPase基因。2008年,Detmer等[21]构建了在周围运动神经元表达人源Mfn2T105M基因的转基因小鼠,该小鼠通过剂量依赖效应表现出典型的CMT2A2的临床特征,其中杂合子小鼠出生时仅表现出轻微的短尾,而纯合子小鼠出生时则出现明显的短尾及畸形,还出现后肢紧缩,不能正常伸展,以及严重的步态异常。为了进一步了解纯合子小鼠后肢缺陷的原因,秤取了该小鼠腓肠肌的重量,发现突变纯合子小鼠腓肠肌的重量明显减轻,肌肉病理检查也证实腓肠肌明显萎缩。但小鼠转轴实验和步态分析未见明显异常。由于Mfn2T105M突变能引起线粒体分布缺陷,为此Detmer等检测了转基因小鼠运动神经元中线粒体的形态学和分布,定量实验证实纯合子小鼠运动神经元中线粒体分布不均匀,出现线粒体异常聚集,推测这是导致轴索病变的原因。值得指出的是,小鼠运动神经元中线粒体分布紊乱及产生的神经病变只发生在过表达的转基因小鼠中,而未出现在基因敲入小鼠中,原因尚在分析中。
3.3 CMT2B1小鼠模型
1999年,Bouhouche等[37]报道了核纤层蛋白A/C(LMNA)基因突变(c.Arg298Cys)可导致CMT2B1。同年 Sullivan等[38]报道了纯合型Lmna敲除小鼠表现出异常步态、后肢疲软、前肢无法支撑整个身体因此行走姿势僵硬,且在生长发育过程中出现进行性加重的脊柱侧弯,但在同窝杂合子小鼠中未观察到上述表型。为了进一步了解LMNA纯合突变对外周轴索损伤的作用,Sullivan等[38]对Lmna基因敲除小鼠的坐骨神经进行了超微结构的分析,结果发现突变纯合子小鼠坐骨神经轴索密度大幅度降低,甚至出现无髓鞘轴索,这些表型都与2002年De Sandre-Giovannoli等[23]报道的 AR-CMT2患者的表型特征极为相似。以往的研究还表明LMNA基因突变与肢带型肌营养不良 1B型(LGMD1B)、AD遗传的Emery-Dreifuss肌营养不良(AD-EDMD)和扩张性心肌病伴心脏传导阻滞 1A(CMD1A)等疾病有关,表明核纤层蛋白A/C的不同功能域对于维持和整合不同细胞系的功能至关重要。因此不同疾病的相似特征在临床诊断分型中需格外注意。
3.4 CMT2D小鼠模型
已知编码甘氨酰-tRNA合成酶基因(GARS)的突变可导致人类CMT2D。2006年,Seburn等[25]通过定位克隆构建了突变诱导的 Nmf249显性小鼠模型。在该小鼠中,Gars基因有一段 AAATA取代了 CC,导致由该基因第 278位编码的脯氨酸(Pro)突变为酪氨酸(Tyr)和赖氨酸(Lys),结果突变纯合子小鼠(GarsNmf249/Nmf249)出现胚胎致死现象,而杂合子小鼠(GarsNmf249/+)则出现运动感觉神经疾病的症状,还在3周龄时出现神经肌肉功能障碍,尤以远端肌肉为甚,体型较小,寿命缩短。病理检查发现轴索直径减小,大直径轴索数量减少,神经肌肉接头(NMJ)的突触连接处及肌肉出现去神经化病理改变,但未发现脱髓鞘现象。神经电生理检查显示突变小鼠的神经传导速度显著降低(约下降 60%)。分子生物学分析显示该突变并未影响Gars基因的mRNA表达,并且对甘氨酰-tRNA合成酶的酶动力学以及酶的活性都没有产生影响。Seburn等[25]认为这些现象不能用单倍体缺失效应或者显性负性效应来解释,因此提出了另一种假设,即GarsNmf249基因突变引起的外周神经病变与甘氨酰-tRNA合成酶(GlyRS)蛋白的功能异常有关,而与GlyRS活性丧失无关。
3.5 CMT2E小鼠模型
人体中神经丝蛋白轻链(NF-L,又称 NEFL)的突变与 CMT2E相关,受累者的严重程度随不同突变而有所不同,且同一突变在不同家庭中也会有所不同,呈现出异质性的特征。为了研究该病的致病机制,Shen等[28]构建了一种新的CMT2E小鼠模型,该小鼠持续表达带有人类hNF-LE397K基因突变的产物。表型分析发现,不同年龄阶段小鼠体内 hNF-L受限程度不同,并表现出进行性加重的现象。其中1月龄hNF-LE397K小鼠以运动神经元胞体(NFs)磷酸化的异位积累为特征,表现出相对较轻的神经病变。4月龄小鼠出现明显的表型,包括异常的后肢姿势,足趾畸形,自发性活动减少,感觉神经的轴索数量减少,运动神经传导速度(MNCVs)降低。有趣的是,随着年龄增大,突变小鼠并未出现 NFs在运动神经元中的大量累积,NF网状结构似乎也没有受到大范围的影响。下肢远端肌肉萎缩,但仍然受坐骨神经的支配,也未去神经化。Shen等[28]推测产生这种现象的原因可能与突变小鼠肌肉的感觉神经支配发生的改变有关,即因大量感觉神经的轴索缺失造成后肢姿势异常。除了hNF-LE397K小鼠模型外,目前还建成了hNF-LP22S小鼠模型[30],两者相比在表型方面较为相似,但在病理机制方面则存在差异,说明不同功能域改变可导致不同的病理变化。
3.6 CMT2F小鼠模型
2001年,Ismailov等[39]首次报道热休克蛋白 β1(HSPB1)基因突变(S135F)可导致人类CMT2F。2011年,d’Ydewalle等[32]建立了人源Hspb1S135F转基因小鼠,表型分析显示该小鼠具有轴索性CMT受累者的临床和病理特征。所有转基因小鼠出生时均正常,符合孟德尔遗传规律,存活率与野生型小鼠相比没有显著差异。6个月后进行悬尾实验发现,Hspb1S135F突变小鼠的后肢出现萎缩病变; 行为学实验(转轴和热板实验)显示突变小鼠出现进行性运动损伤,肌张力降低。6~8月龄小鼠的复合肌肉动作电位(cMAPs)的波幅明显降低,且感觉神经动作电位(SNAPs)中基线到波峰的振幅明显降低。该课题组认为Hspb1S135F小鼠表型可用功能获得性机制解释。研究还发现由Hspb1S135F小鼠中分离的背根神经节(DRG)神经元细胞中线粒体运输受到严重影响。进一步分析显示沿着轴索的胞内运输需动力蛋白在指定的分子(如乙酰化的 α-微管蛋白)作用下运送“货物”。对于线粒体运输而言,移动的线粒体会优先定位于乙酰化的微管处。如果 α-微管蛋白乙酰化过程受阻就容易导致神经退行性疾病的产生。d’Ydewalle 等发现转基因小鼠外周神经中乙酰化的 α-微管蛋白总量大幅降低。但是,抑制微管蛋白去乙酰化酶(HDAC6)可以重新调节乙酰化的 α-微管蛋白水平,缓解轴索运输障碍,表明Hspb1突变导致的 CMT2F 可能存在去乙酰化机制。使用α-微管蛋白乙酰化抑制剂治疗Hspb1突变小鼠,能在行为学与电生理水平部分修复CMT2F的表型,表明α-微管蛋白乙酰化水平的降低在由HSPB1突变导致的CMT2F的致病机制中发挥重要作用。
3.7 CMT2Q小鼠模型
我们对来自山东高密的一个CMT2大家系进行了研究。全基因组扫描和连锁分析显示疾病表型与10p12-14紧密连锁,该区域覆盖 D10S585与D10S1477之间的微卫星标记,共5.41 Mb。DNA序列分析发现所有受累者的脱氢酶 E1和转酮酶结构域 1基因(DHTKD1)第 8外显子存在一个无义突变[c.1455T>G(p.Tyr485*)][19]。进一步研究显示受累者外周血中DHTKD1的mRNA表达水平只有未受累者的一半。细胞水平研究显示在转染了带无义突变的mRNA和截短蛋白的细胞中转录产物的降解速度明显快于野生型细胞。沉默无义介导的 mRNA降解(NMD)途径中的促进因子UPF1能有效地阻止因无义突变所造成的mRNA和蛋白质水平的下降[40]。更为重要的是,沉默DHTKD1可导致细胞能量产生的受限,表现为 ATP、总 NAD+和 NADH,以及NADH水平的下降。综上所述DHTKD1的无义突变是导致CMT2Q的分子基础,提示DHTKD1在线粒体能量产生及神经发育中起着重要作用[19]。目前国内外学者有关DHTKD1基因功能的研究仅限于体外研究,活体内的研究尚未展开,所以对于DHTKD1的功能缺失作用能否在小鼠体内再现尚不明确。为了更好地研究DHTKD1基因的生物学功能及该基因突变导致CMT2Q的分子机制,我们利用Red/ET同源重组技术构建了Dhtkd1点突变基因敲入打靶载体,通过同源重组技术成功地在Dhtkd1基因第8号外显子和第9号外显子之间插入1个Lox P位点、2个Frt及1个neo筛选标记,通过囊胚腔注射获得嵌合体小鼠,并通过与野生型小鼠交配得到含有突变位点的杂合子小鼠。接着,杂合子小鼠互相交配得到突变纯合子、杂合子与野生型小鼠,为下一步从动物水平研究Dhtkd1基因的生物学功能奠定了基础[34]。目前进行的表型分析显示 3种基因型小鼠的繁殖情况正常,野生型、杂合子和纯合子小鼠的比例符合孟德尔遗传定律(1∶2∶1),并未出现纯合子胚胎致死或早亡。3种基因型小鼠坐骨神经的病理学分析(包括半薄切片光镜和电镜观察)显示突变纯合子小鼠轴索中大径纤维的比例明显少于野生型小鼠,且髓鞘出现内陷、外凸或畸形的比例也高于野生型小鼠。实时荧光定量PCR实验结果显示野生型小鼠坐骨神经和肝脏中DHTKD1的转录产物(mRNA)水平最高,突变纯合子最低,杂合子介于二者之间。突变纯合子小鼠肾脏中DHTKD1未检出、杂合子仅为野生型的一半含量; 能量代谢实验结果显示突变纯合子小鼠的ATP含量明显低于野生型,且NADP+/NADPH的比值也存在显著性差异。行为学实验显示突变纯合子小鼠较野生小鼠的感觉迟钝(足爪热痛实验); 野生型小鼠在滚轴上持续的时间高于杂合子和纯合子(滚轴试验)。考虑到在人体中CMT2Q属晚发型、渐进性疾病,并且已有的一些 CMT2型小鼠模型的发病年龄大多在 10月龄以后,所以Dhtkd1Tyr486*敲入小鼠模型可能尚未到出现明显表型的年龄,我们期望随着突变敲入小鼠年龄的增长能观察到更明显的表型。
4 问题与展望
CMT2是一种高度遗传异质性的疾病,目前已克隆了17个致病基因,但仍有一些CMT2的致病基因未知。随着外显子捕获及二代测序技术的日益普及,相信会有更多导致 CMT2的致病基因被定位和克隆[41],并为突变基因致病机制的研究及临床诊治奠定基础。为了从动物水平明确CMT2的发病机制,就必须借助动物模型开展相关研究。目前已成功建立了近 10种 CMT2的动物模型,并能部分模拟CMT2各亚型的临床表型。然而,在小鼠模型构建中也经常会出现小鼠模型与人类疾病表型不一致甚至不出现任何表型的情况,这对研究人员而言是个棘手的问题。产生这种现象的原因是多方面的:其一,小鼠和人类基因组虽然同源性极高,但同源性毕竟未达到100%,尤其在信号传导、生理生化方面仍存在很多差异; 其二,机体发育过程中其他基因的补救或代偿作用可以减弱甚至消除某一突变基因的致病效应; 其三,在基因工程小鼠构建过程中,引入单个点突变的同时也会引入 neo等筛选标记。虽然多数情况下筛选标记的引入对基因功能不会产生显著影响,但有时也会对被修饰基因产生一些负面作用,从而很难实现完全自然状态的模拟[28]。为此,在CMT2小鼠模型建立时必须充分考虑上述因素的影响,并采取相应的应对策略。
已知 CMT2是一种神经源性肌肉疾病,涉及到轴索的长距离运输。由于人类与小鼠在体型及解剖结构方面存在一定的差异,特别是人类轴索延伸至下肢的距离相比小鼠的距离要长,因此突变对小鼠下肢肌肉的影响可能会较小,病变程度也可能较轻。近年来对运动神经元疾病的研究还发现微环境在CMT2发病过程及疾病进展过程中起着至关重要的作用。携带突变基因的运动神经元通常可在衰老及炎症环境的诱导下发生病变,而病变后的运动神经元又反过来使得微环境及胶质细胞更加恶化,这样疾病逐渐发展[42]。为进一步研究CMT2的发病机制,还需要构建更多在时间和空间方面能得到控制的CMT2小鼠模型。
科学工作者开展CMT2研究的目的都是为了人类健康,然而到目前为止,还没有一项研究专门侧重于尚未出现CMT2症状的年轻人的心理健康。国际遗传顾问协会(NSGC)和美国人类遗传协会(ASHG)发起在儿童中检测成年期发病的活动,目的就是为儿童提供更好的照顾。该项活动侧重于病人自发的、符合法律规定的检测,所有先证者和家人都应考虑到医疗、社会、心理和生殖问题。对于CMT而言,无症状或症状不明显的儿童不会从确诊为CMT疾病中获得任何好处,变幻莫测的疾病发展趋势以及缺乏预防保健意识无疑对儿童的未来生涯和人生规划产生巨大挑战。因此早期诊断可以提前阻止疾病的发生并且提供长期的护理保障,这对于儿童未来的生活将会产生巨大益处[7]。
[1]Dyck PJ,Lambert EH.Lower motor and primary sensory neuron diseases with peroneal muscular atrophy.I.Neurologic,genetic and electrophysiologic findings in hereditary polyneuropathies.Arch Neurol,1968,18(6):603–618.
[2]张如旭,郭鹏,任志军,赵国华,刘三妹,刘婷,资晓宏,胡正茂,夏昆,唐北沙.LITAF、RAB7、LMNA 和MTMR2基因在中国人腓骨肌萎缩症患者的突变分析.遗传,2010,32(8):817–823.
[3]Lupski JR,Reid JG,Gonzaga-Jauregui C,Rio Deiros D,Chen DC,Nazareth L,Bainbridge M,Dinh H,Jing C,Wheeler DA,McGuire AL,Zhang F,Stankiewicz P,Halperin JJ,Yang C,Gehman C,Guo D,Irikat RK,Tom W,Fantin NJ,Muzny DM,Gibbs RA.Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy.N Engl J Med,2010,362(13):1181–1191.
[4]Pareyson D,Scaioli V,Laurà M.Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease.Neuromolecular Med,2006,8(1–2):3–22.
[5]Banchs I,Casasnovas C,Albertí A,De Jorge L,Povedano M,Montero J,Martínez-Matos JA,Volpini V.Diagnosis of Charcot-Marie-Tooth disease.J Biomed Biotechnol,2009,2009:985415.
[6]Harding AE,Thomas PK.The clinical features of hereditary motor and sensory neuropathy types I and II.Brain,1980,103(2):259–280.
[7]Siskind CE,Panchal S,Smith CO,Feely SM,Dalton JC,Schindler AB,Krajewski KM.A review of genetic counseling for Charcot Marie Tooth Disease(CMT).J Genet Couns,2013,22(4):422–436.
[8]Bird TD.Charcot-Marie-Tooth Neuropathy Type 2.Gene-Reviews,http://www.ncbi.nlm.nih.gov/books/NBK1285,2013/09/03.
[9]徐汪洋,孙莲花,顾鸣敏.腓骨肌萎缩症的遗传特征及致病机制的研究进展.国际遗传学杂志,2010,33(2):93–96,124.
[10]Patzkó Á,Shy ME.Update on Charcot-Marie-Tooth disease.Curr Neurol Neurosci Rep,2011,11(1):78–88.
[11]Züchner S,Vance JM.Molecular genetics of autosomaldominant axonal Charcot-Marie-Tooth disease.Neuromolecular Med,2006,8(1–2):63–74.
[12]顾鸣敏,王铸钢.《医学遗传学》(第3版).上海:上海科学技术文献出版社,2013,75–80.
[13]傅继梁主编,王铸钢副主编.基因工程小鼠.上海:上海科学技术出版社,2006,5–18.
[14]吴乃虎,张方,黄美娟.基因工程术语.北京:科学出版社,2006,193–194,373.
[15]张付峰.HSP22转基因小鼠模型的建立[学位论文].长沙:中南大学,2008:46–72.
[16]胡以平,曾溢滔.后基因组时代的基因工程小鼠.第二军医大学学报,2003,24(2):117–119.
[17]Zhao C,Takita J,Tanaka Y,Setou M,Nakagawa T,Takeda S,Yang HW,Terada S,Nakata T,Takei Y,Saito M,Tsuji S,Hayashi Y,Hirokawa N.Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bβ.Cell,2001,105(5):587–597.
[18]Saporta MA,Shy BR,Patzko A,Bai Y,Pennuto M,Ferri C,Tinelli E,Saveri P,Kirschner D,Crowther M,Southwood C,Wu X,Gow A,Feltri ML,Wrabetz L,Shy ME.MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-Tooth disease type 1B.Brain,2012,135(7):2032–2047.
[19]Xu WY,Gu MM,Sun LH,Guo WT,Zhu HB,Ma JF,Yuan WT,Kuang Y,Ji BJ,Wu XL,Chen Y,Zhang HX,Sun FT,Huang W,Huang L,Chen SD,Wang ZG.A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth disease type 2 in a large Chinese pedigree.Am J Hum Genet,2012,91(6):1088–1094.
[20]Tanaka Y,Hirokawa N.Mouse models of Charcot-Marie-Tooth disease.Trends Genet,2002,18(12):S39–S44.
[21]Detmer SA,Vande Velde C,Cleveland DW,Chan DC.Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A.Hum Mol Genet,2008,17(3):367–375.
[22]Guillet V,Gueguen N,Cartoni R,Chevrollier A,Desquiret V,Angebault C,Amati-Bonneau P,Procaccio V,Bonneau D,Martinou JC,Reynier P.Bioenergetic defect associated with mKATP channel opening in a mouse model carrying a mitofusin 2 mutation.FASEB J,2011,25(5):1618–1627.
[23]De Sandre-Giovannoli A,Chaouch M,Kozlov S,Vallat JM,Tazir M,Kassouri N,Szepetowski P,Hammadouche T,Vandenberghe A,Stewart CL,Grid D,Lévy N.Homozygous Defects in LMNA,encoding lamin A/C Nuclear-Envelope Proteins,cause autosomal recessive axonal neuropathy in human(Charcot-Marie-Tooth Disorder Type 2) and mouse.Am J Hum Genet,2002,70(3):726–736.
[24]Poitelon Y,Kozlov S,Devaux J,Vallat JM,Jamon M,Roubertoux P,Rabarimeriarijaona S,Baudot C,Hamadouche T,Stewart CL,Levy N,Delague V.Behavioral and molecular exploration of the AR-CMT2A mouse model Lmna(R298C/R298C).Neuromolecular Med,2012,14(1):40–52.
[25]Seburn KL,Nangle LA,Cox GA,Schimmel P,Burgess RW.An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model.Neuron,2006,51(6):715–726.
[26]Motley WW,Seburn KL,Nawaz MH,Miers KE,Cheng J,Antonellis A,Green ED,Talbot K,Yang XL,Fischbeck KH,Burgess RW.Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wildtype GARS levels.PLoS Genet,2011,7(12):e1002399.
[27]Seo AJ,Shin YH,Lee SJ,Kim D,Park BS,Kim S,Choi KH,Jeong NY,Park C,Jang JY,Huh Y,Jung J.A novel adenoviral vector-mediated mouse model of Charcot-Marie-Tooth type 2D(CMT2D).J Mol Histol,2013,Epub ahead of print.
[28]Shen H,Barry DM,Dale JM,Garcia VB,Calcutt NA,Garcia ML.Muscle pathology without severe nerve pathology in a new mouse model of Charcot-Marie-Tooth disease type 2E.Hum Mol Genet,2011,20(13):2535–2548.
[29]Dale JM,Villalon E,Shannon SG,Barry DM,Markey RM,Garcia VB,Garcia ML.Expressing hNF-LE397K results in abnormal gaiting in a transgenic model of CMT2E.Genes Brain Behav,2012,11(3):360–365.
[30]Filali M,Dequen F,Lalonde R,Julien JP.Sensorimotor and cognitive function of a NEFL(P22S) mutant model of Charcot-Marie-Tooth disease type 2E.Behav Brain Res,2011,219(2):175–180.
[31]Dequen F,Filali M,Larivière RC,Perrot R,Hisanaga S,Julien JP.Reversal of neuropathy phenotypes in conditional mouse model of Charcot-Marie-Tooth disease type 2E.Hum Mol Genet,2010,19(13):2616–2629.
[32]d'Ydewalle C,Krishnan J,Chiheb DM,Van Damme P,Irobi J,Kozikowski AP,Vanden Berghe P,Timmerman V,Robberecht W,Van Den Bosch L.HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease.Nat Med,2011,17(8):968–974.
[33]Bogdanik LP,Sleigh JN,Tian C,Samuels ME,Bedard K,Seburn KL,Burgess RW.Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot-Marie-Tooth disease.Dis Model Mech,2013,6(3):780–792.
[34]郭文婷.Dhtkd1基因点突变敲入小鼠模型的建立与表型初步分析[学位论文].上海:上海交通大学,2013.
[35]Saito M,Hayashi Y,Suzuki T,Tanaka H,Hozumi I,Tsuji S.Linkage mapping of the gene for Charcot-Marie-Tooth disease type 2 to chromosome 1p(CMT2A) and the clinical features of CMT2A.Neurology,1997,49(6):1630–1635.
[36]Züchner S,Mersiyanova IV,Muglia M,Bissar-Tadmouri N,Rochelle J,Dadali EL,Zappia M,Nelis E,Patitucci A,Senderek J,Parman Y,Evgrafov O,Jonghe PD,Takahashi Y,Tsuji S,Pericak-Vance MA,Quattrone A,Battaloglu E,Polyakov AV,Timmerman V,Schröder JM,Vance JM.Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A.Nat Genet,2004,36(5):449–451.
[37]Bouhouche A,Benomar A,Birouk N,Mularoni A,Meggouh F,Tassin J,Grid D,Vandenberghe A,Yahyaoui M,Chkili T,Brice A,LeGuern E.A locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 1q21.2-q21.3.Am J Hum Genet,1999,65(3):722–727.
[38]Sullivan T,Escalante-Alcalde D,Bhatt H,Anver M,Bhat N,Nagashima K,Stewart CL,Burke B.Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy.J Cell Biol,1999,147(5):913–920.
[39]Ismailov SM,Fedotov VP,Dadali EL,Polyakov AV,Van Broeckhoven C,Ivanov VI,De Jonghe P,Timmerman V,Evgrafov OV.A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2(CMT2F) maps to chromosome 7q11-q21.Eur J Hum Genet,2001,9(8):646–650.
[40]郭文婷,徐汪洋,顾鸣敏.无义介导的mRNA降解机制及其在单基因遗传病中的作用.遗传,2012,34(8):935–942.
[41]Weedon MN,Hastings R,Caswell R,Xie W,Paszkiewicz K,Antoniadi T,Williams M,King C,Greenhalgh L,Newbury-Ecob R,Ellard S.Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease.Am J Hum Genet,2011,89(2):308–312.
[42]Azzedine H,Senderek J,Rivolta C,Chrast R.Molecular genetics of charcot-marie-tooth disease:from genes to genomes.Mol Syndromol,2012,3(5):204–214.
