利用改进的MutMap方法克隆水稻雄性不育基因
2014-11-17陈竹锋严维王娜张文辉谢刚卢嘉威简智华刘东风唐晓艳
陈竹锋,严维,王娜,张文辉,谢刚,卢嘉威,简智华,刘东风,唐晓艳,
1.深圳市作物分子设计育种研究院,深圳 518107;2.首都师范大学生命科学学院,北京 100089
雄性不育是影响水稻产量的关键因素之一,但在另一方面,为了提高产量和获取杂种优势,在杂交育种过程中,雄性不育又具有非常重要的应用价值[1,2]。在水稻杂交制种过程中,雄性不育系的利用降低了生产成本,提高了制种产量,充分发挥了杂交的优势。同时,雄性不育又是研究水稻花发育分子调控机理的重要研究材料[3~6]。因此,开展水稻育性基因克隆不仅具有生产利用价值,而且具有重要的理论意义。
突变体是功能基因组学研究的重要材料,近年来,利用水稻突变体进行水稻功能基因组学研究取得重大进展[7~11]。分离克隆相关基因是研究利用突变体的前提和基础,目前水稻突变体基因克隆最常用的技术有图位克隆(Positional cloning)、转座子标签(Transposon tagging)、T-DNA 插入(T-DNA tagging)和反向遗传学(Reverse genetics)等技术。
但是,随着各水稻基因组测序的完成以及二代测序技术的日趋成熟,MutMap克隆基因方法应运而生[12],该方法综合运用全基因组学、生物信息学、分子遗传学和分子生物学等多学科交叉研究方法,结合高通量的二代测序方法,充分利用水稻重测序的数据及分析优势,通过把突变体群体重测序结果与完全组装的野生型基因组序列相比对,找出水稻突变体基因。该方法大大降低了基因克隆成本,缩短了基因克隆时间。
本研究以 EMS(甲磺酸乙酯)诱变水稻品种黄华占得到的一个雄性不育突变体为研究材料,命名为osms55,对其进行初步的表型鉴定、遗传分析及遗传背景鉴定,并利用改进的 MutMap方法,结合HRM及cDNA序列比对,成功克隆了该雄性不育基因。
1 材料和方法
1.1 材料
水稻雄性不育突变体osms55是本实验室用EMS诱变籼稻黄华占(HHZ)种子(处理浓度为 0.7%,处理时间12 h),进一步筛选M2突变体库而获得,该突变体植株与野生型黄华占植株杂交,产生的 F1和F2代植株用于表型分析和遗传分离分析。F2代的雄性不育植株用于MutMap 分析和基因克隆。所有植物材料种植于深圳作物分子设计育种研究院光明试验基地。
1.2 方法
1.2.1 花粉育性观察及表型鉴定
采用I2-KI染色方法鉴定水稻花粉育性。取成熟的水稻颖花,用镊子将 6枚花药取出置于载玻片上,用镊子小心将花药夹碎后浸于适量的1% I2-KI染液中 1~2 min,去除花药壁等残留物后置于光学显微镜下观察并拍照。
1.2.2 突变体遗传背景鉴定
采用基因组 DNA提取试剂盒(Qiagen),抽提突变体与野生型材料基因组 DNA,采用 Illumina Infinium iSelect SNP(50 K)芯片检测技术[13],对比分析野生型黄华占与突变体的SNP位点差异,分析判断突变体是否为黄华占来源。
1.2.3 不育基因克隆
将突变体材料与野生型材料杂交获得 F1个体,F1自交获得F2分离群体,种植F2群体,并从F2群体中挑选30株具有雄性不育表型的植株,取其叶片提取 DNA,等量混合形成 DNA pool,采用改进的MutMap方法[12]克隆不育基因,并获得基因序列和突变位点信息。
突变体DNA pool用超声波方法随机打断,依照Illumina Paired-End DNA Sample Prep kit 建议的方法建库。然后选取长度为 200~300 bp之间的片段,采用Hiseq 2000平台(PE101)进行重测序。测序原始数据经过质量监控,去除低质量及接头污染的数据后,通过SOAP2软件[14],将数据比对到日本晴参考基因组上,得到比对到唯一位置的reads。然后利用这些数据,采用SOAPsnp软件[15]寻找突变体与日本晴参考基因组间的单核苷酸多态性(SNP)。
采用同样的方法测序并分析野生型 HHZ个体重测序数据,得到野生型HHZ与日本晴之间的SNP位点。然后将osms55/日本晴 SNP与野生型 HHZ/日本晴 SNP相比较,去掉突变体中与野生型相同基因型的 SNP位点后,并根据野生型 HHZ Allele index≥0.8(唯一比对到该位点且支持野生型基因型的 reads数与所有覆盖到该位置的 reads数的比值)以及覆盖到该位点的reads≥5进行筛选。计算筛选剩下的位点的 SNP index值,并将所有筛选剩下的位点在染色体上作图。理论上,造成突变性状的突变位点SNP index应该等于1,即为纯合位点; 由于遗传连锁,其附近的 SNP位点 index应该等于或接近 1。寻找在染色体上成簇分布的 SNP位点,再根据EMS诱变通常产生的突变,即G→A或 C→T突变,以及突变位点对所在基因的影响加以判断,确定用于实验验证的突变位点。改进的 MutMap方法将突变体群体测序结果和野生型基因组序列结果分别与日本晴参考基因组相比对,利用 SNP index=1和差异位点分布等指标找出候选突变基因。
1.2.4 基因型测定分型SNP
采用高分辨率溶解方法(High Resolution Melting,HRM)[16,17]鉴定 F2个体的基因型,将测序后已知基因型的材料作为对照,根据突变基因序列设计PCR引物(表1)。将PCR产物转移至Light Scanner中进行扫描分型,通过荧光信号变化和自动分型软件绘制溶解曲线,软件根据荧光值及曲线准确将检测样品区分为野生纯合型、杂合型、突变纯合型。
1.2.5 RNA提取与反转录
样品组织在液氮中进行研磨后,采用 TRIzol(Invitrogen)法提取小穗中的总 RNA,采用 Super-Script®Ⅲ First Strand Synthesis Kit试剂盒(Invitrogen)反转录得到 cDNA样品。反转录产物作为模板,通过PCR 方法扩增野生型和突变体cDNA,用作序列分析。

表1 用于HRM分析的引物及序列
2 结果与分析
2.1 osms55突变体表型鉴定
雄性不育突变体osms55在营养生长期与野生型黄华占相比,植株表型上没有明显的差异。材料授粉灌浆25 d后,野生型黄华占结实率在85%以上,叶片逐渐衰老; 而osms55不结实,谷粒空瘪,植株大部分叶片仍然青绿(图1:a、b)。解剖观察黄华占和突变体osms55成熟颖花,发现两者花药形态无明显差异,突变体雌蕊外观正常(图1:c、d)。碘染结果表明突变体osms55花粉不能正常染色,以碘败型花粉为主(图1:e、f)。
2.2 osms55突变体的遗传分析
突变体osms55与野生型的黄华占杂交,其 F1代植株花粉育性表现为正常,小穗结实率89.8%。F2代植株群体可明显的区分为正常植株与不育植株两组,在 1 351株的植株里,正常可育植株 1 020株,不育植株 331株,经卡方(χ2)测验,其分离比符合3∶1(表 2),表明osms55的突变性状由单隐性核基因控制,将该基因命名为OsMS55。
2.3 突变体osms55的遗传背景鉴定
利用Illumina Infinium iSelect SNP(50 K)芯片检测技术[13]比较野生型黄华占与突变体osms55之间的SNP位点差异,鉴定突变体的遗传背景。该芯片共有51,599个SNP位点,均匀分布在水稻的12条染色体上。除去未检测到的位点,该方法共检测到出48,040个SNP位点; 其中突变体与野生型黄华占不同的位点有692个,其中686个为杂合位点,仅有6个纯合位点与野生型黄华占不同,差异位点集中分布在特定的染色体区域(图2)。初步判断该突变体的遗传背景与黄华占一致,突变体材料来源于黄华占。检测到的这些差异位点可能基于以下两方面的原因:(1)传统育种方法培育的水稻品种来源姊妹系混合的小群体,而非单株遗传;(2)自交繁殖世代相对较低。

图1 突变体的表型特征

表2 突变体osms55与HHZ杂交F2代的表型分离
2.4 突变体osms55的基因克隆
突变体 DNA pool通过重测序,共得到 76 658 101 PE reads,采用SOAP2软件[14]将数据比对到日本晴参考基因组上(http://rice.plantbiology.msu.edu/),覆盖整个基因组89.53%,覆盖深度为30×,比对到唯一位置的reads有90 071 940。利用这些比对到唯一位置的数据,采用SOAPsnp软件寻找与日本晴之间的单核苷酸多态性(SNP),共得到 1 146 971个SNP。采用同样的方法测序野生型HHZ植株基因组,共获得60 726 573 PE reads,reads长度为44 bp,覆盖整个基因组93.35%,覆盖深度为13×,与日本晴基因组比对得到710 055 SNP位点。

图2 osms55的遗传背景鉴定
将野生型 HHZ/日本晴 SNP与突变体osmm55/日本晴SNP位点进行比较,去掉突变体中与野生型相同基因型的位点后,根据野生型黄华占 Allele index≥0.8以及覆盖到的 reads≥5进行筛选,共得到1 732 SNP位点,分布在水稻12条染色体的不同区域上(图 3)。其中 29个位点在突变体中的 SNP index≥0.8,仅有 11个位于基因上(表3),其中某些位点可能是由于测序深度低或测序过程中产生的错误而导致的假阳性位点。
LOC_Os03g23180、LOC_Os04g31310、LOC_Os08g21860、LOC_Os11g20239四个候选基因的突变位点位于内含子区域,LOC_Os01g25910、LOC_Os03g23180、LOC_Os08g02000、LOC_Os10g20980四个基因均为反转录转座子或转座子,LOC_Os07g16950位于第3个exon结束前2 bp,且为同义突变。
根据重测序数据分析,挑选成簇分布且含有位于基因上造成氨基酸或基因变异的 SNP,仅有 2号染色体和 11号染色体上有符合的位点,分别位于LOC_Os02g40450和 LOC_Os11g18290基因上。LOC_Os11g18290基因功能不明,其突变位于距离起始密码子260 bp处,碱基C突变为T,导致该氨基酸由脯氨酸变为亮氨酸。而LOC_Os02g40450基因编码一个DNA解旋酶,参与减数分裂过程中染色体的交叉互换过程[18],其突变位于基因第 4个内含子的剪切识别位点上,碱基G突变为A(图4)。
2.5 利用HRM方法分析突变位点与性状的遗传连锁关系
根据表3所列的候选基因突变位点变化及候选基因相关生物信息学分析,将LOC_Os02g40450、LOC_Os11g18290作为主要候选基因进行实验验证。随机挑取11个可育植株与85个不育植株,利用表1中所设计的引物对 LOC_Os02g40450、LOC_Os11g18290进行 PCR扩增,采用 LightScanner软件分析数据,获得SNP分型结果,统计结果如表4所示。
LOC_Os11g18290突变位点与不育表型不共分离,即突变体osms55的不育表型不是由该基因突变引起的; 而 LOC_Os02g40450突变位点与不育表型共分离,不育植株中该基因位点均发生突变,可育植株中该基因位点为纯合野生型或杂合型,由此推断,osms55突变体产生雄性不育是由于 LOC_Os02g40450基因在剪切识别位点处发生了突变,影响了RNA的正常剪切加工。
2.6 OsMS55基因cDNA序列比对分析
LOC_Os02g40450的突变位于第四内含子的剪切识别位点 GT-AG上,其中 AG的G突变为 A(图4),为检测突变体cDNA序列是否发生变异,提取野生型黄华占和突变体的总RNA,反转录获得cDNA,设计引物(5′-cttcagtgacat tgctctggtc-3′和 5′-cagtttgtgaaaggcactgtgc-3′)并 PCR扩增后测序,结果表明,突变体osms55的剪切识别位点发生变异后,OsMS55基因第5外显子的前15 bp变为内含子,导致突变体蛋白比野生型少5个氨基酸。Blast分析发现LOC_Os02g40450基因与MER3基因同源,已有报道表明,MER3基因在5’UTR区及起始密码子处的大片段缺失可导致雄性不育[18]。
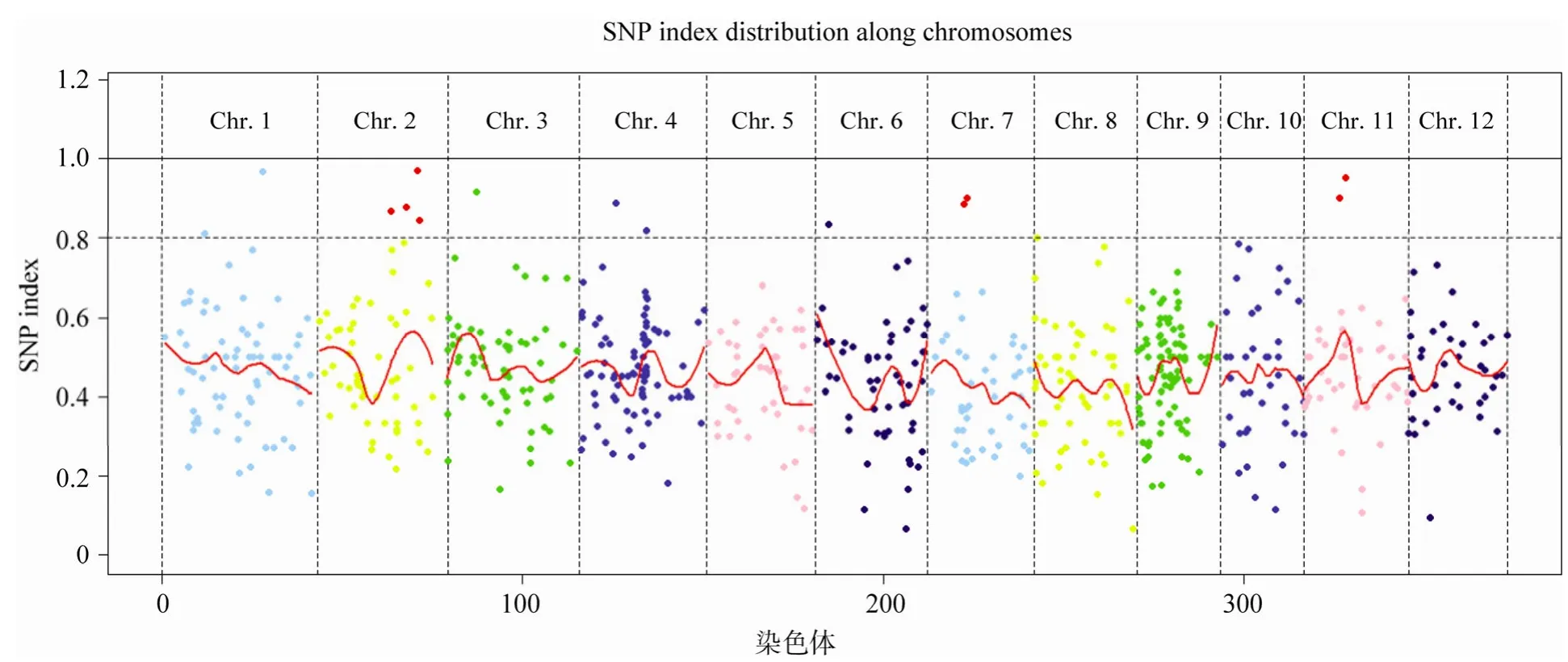
图3 1 732个SNP位点在12条染色体上的分布情况
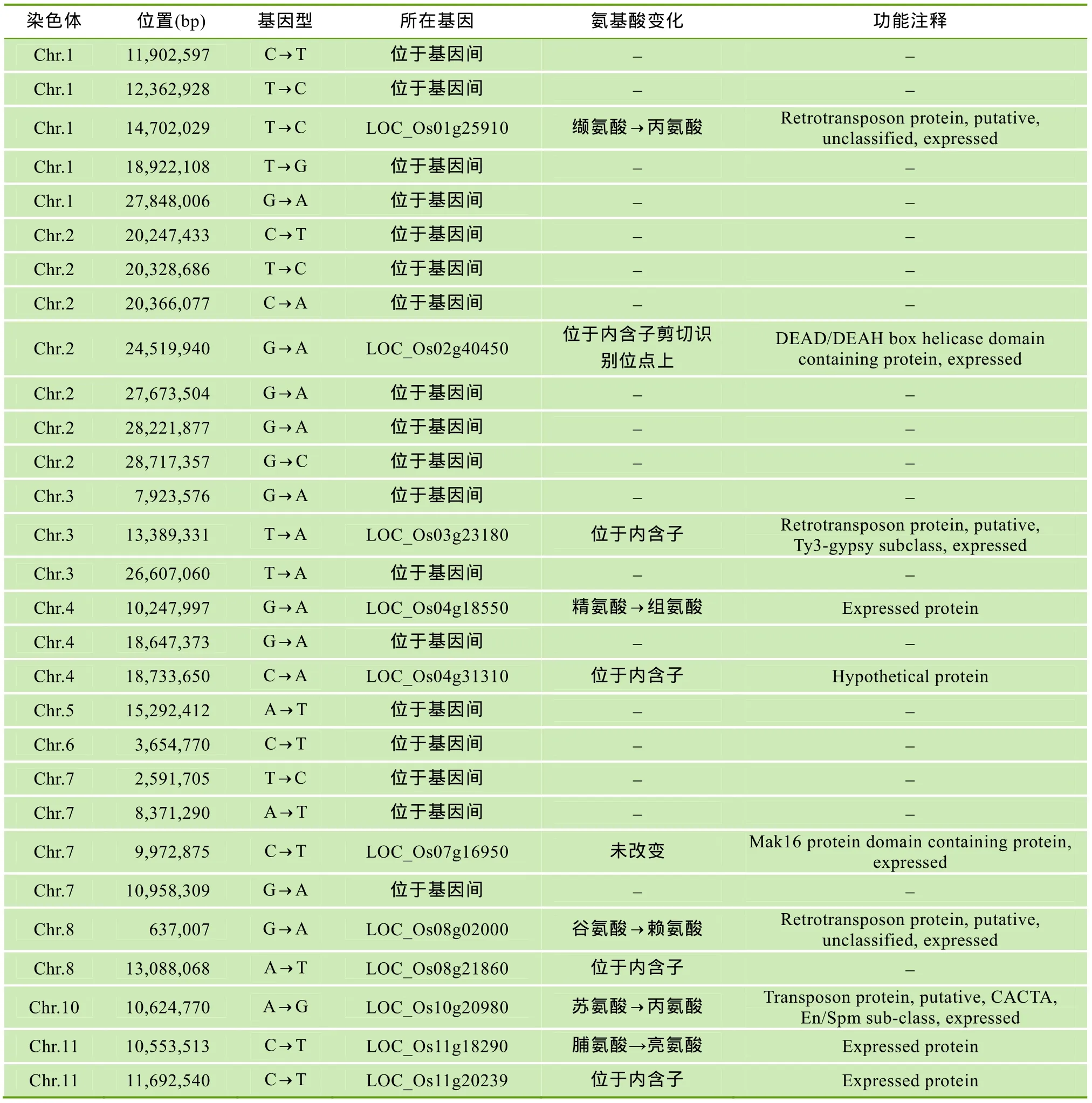
表3 29个SNP index≥0.8的突变区域情况及功能注释
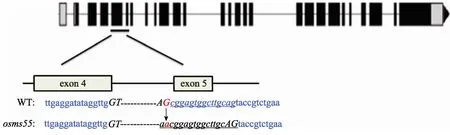
图4 OsMS55基因突变前后序列比对
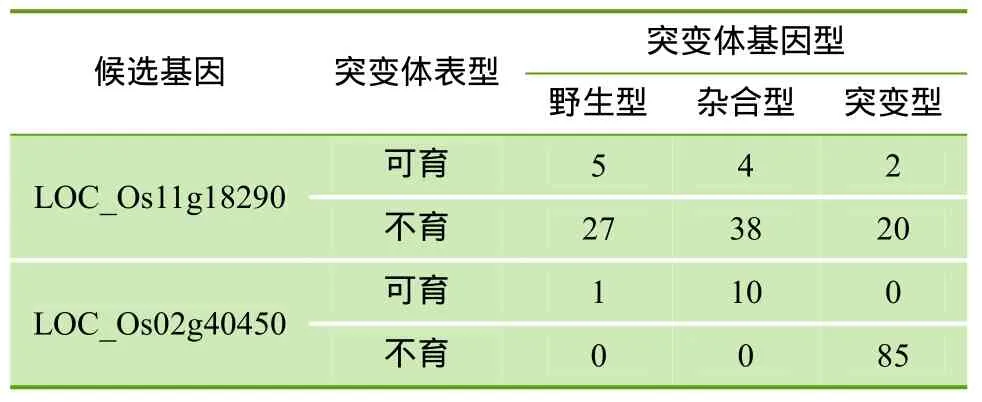
表4 候选基因SNP分型统计结果
3 讨 论
植物的雄性生殖发育是一个多步骤的复杂过程,包括雄蕊分生组织分化,造孢细胞的产生和分化,减数分裂,小孢子母细胞成熟,开花及授粉等,这其中任何一个环节的基因发生突变,均能引起雄器官发育异常,最终产生雄性不育[19,20]。转录组分析表明,约 2.9万个转录本在水稻的花药发育和花粉形成的不同阶段特异表达[21]。到目前为止,涉及水稻花药发育和花粉形成的一些关键基因已被鉴定和研究,这些基因主要参与调控小孢子母细胞发育,绒毡层发育,花粉囊和花粉外壁发育等过程[22~26]。
MutMap方法是Abe等[12]2012年提出的一种基因克隆新方法,相较于图位克隆方法,MutMap方法克隆基因更快捷、效率更高、成本更低,该方法是以二代测序技术为基础,通过对 F2分离群体中隐性表型个体的DNA pool的深度测序,然后把突变体群体测序结果与野生型基因组序列相比对,找出造成突变体性状的基因。MutMap方法不仅可以快速克隆质量性状的隐性核基因,还可以应用到数量性状的QTL 定位[27]。
MutMap方法对于克隆水稻育性发育相关基因尤其具有优势,传统图位克隆方法为了开发更多的分子标记,一般都采用籼粳杂交构建定位分离群体,由此容易产生杂种不育而影响表型判断,而利用回交等方法则增加基因克隆时间与工作量; MutMap只需将突变体与野生型材料杂交一次,自交获得 F2分离群体即可,既简化了群体构建的工作,又能准确判断材料表型。
本研究在Abe等[12]发表的MutMap方法的基础上进行了改进,不再把突变体群体测序结果与野生型基因组序列直接比对,而是把突变体群体测序结果和野生型基因组序列结果分别与日本晴参考基因组相比对,找出各自比对的 SNP位点后,再把两组SNP位点相比对,找出差异位点; 然后再利用 SNP index=1和差异位点分布等指标找出候选突变基因。与 Abe等人的方法相比,本研究所采用的方法不要求精确组装的野生型基因组序列作对照,只需要对突变体群体和野生型个体做 20~30×深度的重测序,即可通过与同一参考基因组相比对,找出候选基因,大大降低了野生型基因组测序和组装成本。
利用改进的 MutMap方法,本研究成功地克隆到雄性不育osms55突变体的基因LOC_Os02g40450,该基因编码的蛋白命名为 MER3[18],该基因突变后减数分裂染色体交叉(CO)数目显著下降,而存在的少数 CO也为随机分布,从而导致水稻产生雄性不育。在Wang等[18]的研究中,mer3突变体中在5’UTR区及起始密码子处有763 bp的缺失,导致MER3基因与其上游基因融合,最终因 MER3蛋白不能正确表达而产生雄性不育。本研究中osms55突变体的突变位点与 Wang等研究不同,是由于突变体基因内含子剪切识别位点发生变异,该变异导致内含子剪切异常,从而产生雄性不育。
目前,水稻隐性雄性核不育材料主要应用于作物育种的群体改良上,实现优质基因叠加,优化育种材料的遗传背景,从而培育出优良新品种或育种亲本[28]。此外,于2009年立项启动实施了“水稻智能不育分子设计技术研究及新型不育系的创制”重点项目,开创了水稻隐性核雄性不育基因在杂交育种上利用的新局面(http://www.most.gov.cn/kjbgz/201009/t20100920_82153.htm)。
智能不育分子设计技术利用遗传稳定的隐性雄性核不育材料,通过转入野生型基因恢复不育材料的花粉育性,同时使用杀花粉基因使含转基因成分的花粉失活,并利用荧光基因分离筛选不育系与保持系种子[29]。该体系不仅有效解决了水稻隐性核雄性不育材料自我繁殖难题,建立了一套高效的杂交水稻育种和制种体系,开创了新的杂交育种途径,拓宽了水稻杂种优势的利用空间,而且能够解决常规杂交育种过程中资源利用率低、育种周期长等瓶颈问题,提高杂交制种纯度,降低杂交制种成本,同时也有效规避转基因生物安全等问题。智能不育技术有效的将现代生物技术与传统育种方法相结合,使得大量水稻隐性雄性核不育基因及突变体材料的利用成为了可能。
本研究中的水稻突变体材料osms55所携带的隐性核不育基因不受光温条件的影响,花粉以碘败为主,雌蕊正常,任何常规品种均可以作为其恢复系,该突变体以及控制该突变体性状的基因为构建新型杂交育种体系提供了必要的元件,克隆该突变基因,为今后的水稻分子设计育种提供更多可供选择的基因及种质资源。
[1]Li SQ,Yang DC,Zhu YG.Characterization and use of male sterility in hybrid rice breeding.J Integr Plant Biol,2007,49(6):791–804.
[2]Budar F,Pelletier G.Male sterility in plants:occurrence,determinism,significance and use.C R Acad Sci III,2001,324(6):543–550.
[3]Kaul MLH.Male sterility in higher plants.Berlin:Springer Verlag,1988:211–256.
[4]Araya A,Zabaleta E,Blanc V,Begu D,Hemould M,Mouras A,Litvak S.RNA editing in plant mitochondria,cytoplasmic male sterility and plant breeding.Electron J Biotechn,1998,1(1):31–39.
[5]Luo DP,Xu H,Liu ZL,Guo JX,Li HY,Chen LT,Fang C,Zhang QY,Bai M,Yao N,Wu H,Wu H,Ji CH,Zheng HQ,Chen YL,Ye S,Li XY,Zhao XC,Li RQ,Liu YG.A detrimental mitochondrial nuclear interaction causes cytoplasmic male sterility in rice.Nat Genet,2013,45(5):573–577.
[6]Xing YZ,Zhang QF.Genetic and molecular bases of rice yield.Annu Rev Plant Biol,2010,61(1):421–442.
[7]郭龙彪,储成才,钱前.水稻突变体与功能基因组学.植物学通报,2006,23(1):1–13.
[8]Krishnan A,Guiderdoni E,An G,Hsing YI,Han CD,Lee MC,Yu SM,Upadhyaya N,Ramachandran S,Zhang Q,Sundaresan V,Hirochika H,Leung H,Pereira A.Mutant resources in rice for functional genomics of the grasses.Plant Physiol,2009,149(1):165–170.
[9]Wang NL,Long T,Yao W,Xiong LZ,Zhang QF,Wu CY.Mutant resources for the functional analysis of the rice genome.Mol Plant,2013,6(3):596–604.
[10]Yang Y,Li Y,Wu CY.Genomic resources for functional analyses of the rice genome.Curr Opin Plant Biol,2013,16(2):157–163.
[11]Jiang SY,Ramachandran S.Natural and artificial mutants as valuable resources for functional genomics and molecular breeding.Int J Biol Sci,2010,6(3):228–251.
[12]Abe A,Kosugi S,Yoshida K,Natsume S,Takagi H,Kanzaki H,Matsumura H,Yoshida K,Mistsuoka C,Muluneh T,Innan H,Cano L,Kamoun S,Teraushi R.Genome sequencing reveals agronomically important loci in rice using MutMap.Nat Biotechnol,2012,30(2):174–178.
[13]Chen HD,Xie WB,He H,Yu HH,Chen W,Li J,Yu RB,Yao Y,Zhang WH,He YQ,Tang XY,Zhou FS,Deng XW,Zhang QF.A high-density SNP genotyping array for rice biology and molecular breeding.Mol Plant,2013,Epub ahead of print.
[14]Li RQ,Yu C,Li YR,Lam TW,Yiu SM,Kristiansen K,Wang J.SOAP2:an improved ultrafast tool for short read alignment.Bioinformatics,2009,25(15):1966–1967.
[15]Li RQ,Li YR,Fang XD,Yan HM,Wang J,Wang J.SNP detection for massively parallel whole-genome resequencing.Genome Res,2009,19(6):1124–1132.
[16]Liew M,Seipp M ,Durtschi J,Margraf RL,Dames S,Erali M,Voelkerding K,Wittwer C.Closed-tube SNP genotyping without labeled probes-a comparison between unlabeled probe and amplicon melting.Am J Clin Pathol,2007,127(3):341–348.
[17]Zhou L,Wang L,Palais R,Pryor R,Wittwer CT.Highresolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution.Clin Chem,2005,51(10):1770–1777.
[18]Wang KJ,Tang D,Wang M,Lu JF,Yu HX,Liu JF,Qian BX,Gong ZY,Wang X,Chen JM,Gu MH,Cheng ZK.MER3 is required for normal meiotic crossover formation,but not for presynaptic alignment in rice.J Cell Sci,2009,122(12):2055–2063.
[19]Glover J,Grelon M,Craig S,Chaudhury A,Dennis E.Cloning and characterization of MS5 from Arabidopsis,a gene critical in male meiosis.Plant J,1988,15(3):345–356.
[20]谭何新,文铁桥,张大兵.水稻花粉发育的分子机理.植物学通报,2007,24(3):330–339.
[21]Guo JX,Liu YG.Molecular control of male reproductive devlopment and pollen fertility in rice.J Integr Plant Biol,2012,54(12):967–978.
[22]Yoshida H,Nagato Y.Flower development in rice.J Exp Bot,2011,62(14):4719–4730.
[23]Zhang DB,Luo X,Zhu L.Cytological analysis and genetic control of rice anther development.J Genet Genomics,2011,38(9):379–390.
[24]Zhang H,Liang WQ,Yang XJ,Luo X,Jiang N,Ma H,Zhang DB.Carbon Starved Anther encodes a MYB do-main protein that regulates sugar partitioning required for rice pollen development.Plant Cell,2010,22(3):672–689.
[25]Qin P,Tu B,Wang YP,Deng LC,Quilichini TD,Li T,Wang H,Ma BT,Li SG.ABCG15 encodes an ABC transporter protein,and is essential for post meiotic anther and pollen exine development in rice.Plant Cell Physiol,2013,54(1):138–154.
[26]Niu NN,Liang WQ,Yang XJ,Jin WL,Wilson Z A,Hu JP,Zhang DB.EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice.Nat Commun,2013,4:1445.
[27]Takagi H,Abe A,Yoshida K,Kosugi S,Natsume S,Mitsuoka C,Uemura A,Utsushi H,Tamiru M,Takuno S,Innan H,Cano LM,Kamoun S,Terauchi R.QTL-seq:rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations.Plant J,2013,74(1):174–183.
[28]汪旭东,周开达,李仕贵,黎汉云,高克铭.利用隐性核不育性进行水稻轮回育种初步研究.西南农业学报,2001,14(3):102–106.
[29]邓兴旺,王海洋,唐晓艳,周君莉,陈浩东,何光明,陈良碧,许智宏.杂交水稻育种将迎来新时代.中国科学(生命科学),2013,43(10):864–868.
