基于溶胶凝胶技术的在线固相萃取与高效液相色谱联用测定饮用水中的雌激素残留
2014-10-22李龙飞石晓蕾王亚娜王玟玟何金兴
李龙飞,苏 敏,石晓蕾,王亚娜,王玟玟,何金兴
(齐鲁工业大学食品与生物工程学院,山东 济南 250353)
雌激素是一类化学结构相似、分子中含有18个碳原子的类固醇激素[1],该类物质可通过模拟或拮抗正常的内源性激素,干扰内分泌系统功能,进而对生殖系统、神经系统和免疫系统等产生影响。己烷雌酚(HEX)、己烯雌酚(DES)和双烯雌酚(DS)属于人工合成的雌激素,对动物具有显著的促生长作用,曾在多国被作为促生长剂广泛用于畜牧业[2]。但如今越来越多的研究表明雌激素可对人和动物的内分泌系统产生严重干扰,且具有潜在的致癌性[3-6]。鉴于此,美国、欧盟等多国已明文规定对其禁用或限用,我国也在2002年明确规定对其禁用[7]。
溶胶凝胶技术是一种非常有发展前途的材料制备方法,该方法反应条件温和,可用于制备粒径不同、形状各异的无机材料和具有特定官能团的无机-有机杂化材料,所得材料具有稳定性好、选择性高、传质速度快等优点[8,9]。本实验以溶胶凝胶技术合成的聚合物为固相萃取填料,考察制备的固相萃取材料对HEX、DES和DS 3种雌激素的吸附效果。
目前针对兽药残留常用的检测方法有固相萃取-高效液相色谱法(SPE-HPLC)[10,11]、高效液相色谱-质谱联用法(HPLC-MS)[4,12]、气相色谱-质谱联用法(GC-MS)[13,14]等。上述方法的检出限虽能在一定程度上满足一般兽药残留检测的要求,但都有不足之处,如SPE-HPLC因离线富集易造成样品损失,导致测量误差,且完成一次检测耗时较长;HPLC-MS价格昂贵,一般实验室难以推广,不适于日常样品的检测;GC-MS使用时一般需要进行繁琐的样品衍生化处理,耗时费力。本研究以不锈钢在线富集管代替液相色谱进样环,采用在线固相萃取-HPLC法进行分析,建立了同时测定水样中3种雌激素残留的检测方法。
1 实验部分
1.1 仪器与试剂
岛津LC-20AT高效液相色谱仪,SPD-M20A二极管阵列检测器(日本岛津公司);梅特勒AL104电子天平(中国梅特勒-托利多公司)。
DES、DS、HEX(色谱纯,Sigma 公司);乙腈、甲醇、乙醇(色谱纯,天津化学试剂厂);二次去离子水(DDW,18 MΩ·cm,Milli-Q超纯水器制得);四乙氧基硅烷(tetraethoxysilane,TEOS;分析纯)、3-氨基丙基三乙氧基硅烷(3-aminopropyltriethoxysilane,APTES;分析纯)。
标准储备溶液:称取 DES、DS、HEX 各 2 mg,用甲醇定容于10 mL棕色容量瓶中,配成200 mg/L的混合标准储备液,于4℃条件下保存。根据需要,临用时用纯水稀释成相应浓度的标准工作溶液。
1.2 实际水样的采集与预处理
矿泉水于山东济南长清地区超市购得;自来水于山东济南长清地区学校采得。收集的水样经0.45 μm水系滤膜过滤,置于棕色试剂瓶中,于4℃保存。使用时先用0.1 mol/L的盐酸溶液或氢氧化钠溶液调节pH至7.0。
1.3 实验方法
1.3.1 色谱条件的选择
色谱柱 Xterra C18(250 mm ×4.6 mm,5 μm;Waters,USA);流动相为甲醇和1%乙酸水溶液,流速为1.0 mL/min,柱温为38℃,进样量为20 μL,检测波长为230 nm。采用保留时间定性,外标法峰面积定量。
1.3.2 固相萃取柱的制备
将5 mL乙腈注入50 mL具塞的圆底烧瓶中,加入1 mL APTES,磁力搅拌反应30 min;加入2 mL TEOS,继续搅拌20 min;再加入1 mL 0.01 mol/L醋酸,磁力搅拌10 min,置于水浴锅中于60℃下孵化10 h,过滤,用甲醇洗涤,于真空干燥箱中100℃下老化10 h[9]。将所得材料研磨成粉备用。在空的在线富集管内依次填入0.45 μm有机滤膜、石英棉(经10%硝酸活化)、70 mg合成材料、石英棉(经10%硝酸活化)、0.45 μm有机滤膜,以此作为固相萃取柱,用于预富集。
1.3.3 在线预富集步骤
固相萃取在线富集与高效液相色谱法联用装置参见文献[15],测定水样中雌激素残留的过程如下:首先,将高效液相色谱仪的进样阀扳到进样位,在流动注射分析仪的作用下,样品溶液进入装有吸附材料的固相萃取柱中进行富集,废液通过废液管流出。然后,将进样阀扳到注射位,以使流动相流过富集柱,富集的雌激素在流动相的作用下进入到色谱分离柱前的管道中。最后,洗脱完成后将进样阀再次扳到进样位,以进行下一次富集;与此同时分析物在色谱柱中被分离。
2 结果与讨论
2.1 色谱条件的选择
分别以乙腈和1%乙酸水溶液、甲醇和1%乙酸水溶液作为流动相,考察了不同比例流动相对3种雌激素分离效果的影响。经测试,两种有机溶剂与1%乙酸水溶液在一定配比下均可对3种雌激素实现分离,但考虑到乙腈对人体的影响,最终选用甲醇和1%乙酸水溶液作为流动相进行分离。试验结果表明甲醇和1%乙酸水溶液比例为64∶36(v/v)时,3种雌激素在16 min内能够达到完全的基线分离,且峰形对称性良好(见图1)。
2.2 影响雌激素在线富集的因素
为了使合成材料对雌激素具有理想的富集效果,分别对洗脱溶剂、样品溶液pH、上样流速等影响富集效果的因素进行了优化。
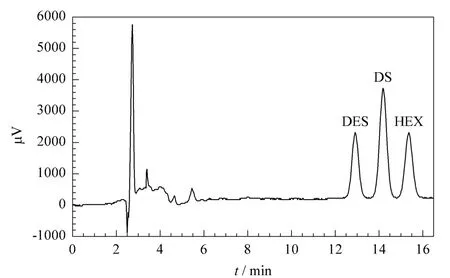
图1 3种雌激素混合标准溶液(1 mg/L)的色谱图Fig.1 Chromatogram of a mixed standard solution of the three estrogens(1 mg/L)
2.2.1 洗脱溶剂的选择
配制10 μg/L的雌激素混合标准溶液进行在线富集,在样品溶液体积为100 mL,上样流速为1 mL/min,洗脱流速为1 mL/min条件下,考察了不同比例的1%乙酸水溶液和甲醇、1%乙酸水溶液和乙腈、1%乙酸水溶液和乙醇作为洗脱溶剂的洗脱效果。结果发现乙醇对于富集在吸附材料上的雌激素几乎没有洗脱效果,而乙腈的洗脱效果明显不如甲醇,由此选择1%乙酸水溶液和甲醇作为洗脱溶剂。考虑到本实验3种雌激素的最佳分离条件,最终选择流动相的配比作为洗脱溶剂的配比。
2.2.2 样品溶液的pH对富集效果的影响
配制 10 μg/L,pH 分别为 5.0、6.0、7.0、8.0、9.0的雌激素混合标准溶液100 mL(采用1 mol/L盐酸溶液调节pH值),控制流速为1 mL/min进行在线富集,洗脱流速同样为1 mL/min,结果如图2所示。从图2可以看出,pH对雌激素的富集效果具有较大的影响,当pH在5.0~7.0时,3种雌激素的峰面积均随pH的升高而增大;而当pH超过7.0时,峰面积显著下降,可能的原因是所合成的固相萃取填料是一种中等极性的化合物,在酸性或中性条件下,有利于填料与雌激素之间氢键的形成,因而具有较高的吸附性。由此最终选择样品溶液的pH为7.0用于后续的试验。
2.2.3 上样流速对富集效果的影响
对100 mL 10 μg/L的雌激素混合标准溶液在线富集,调节蠕动泵的转速,逐步增加上样速度,以考察上样流速对3种雌激素吸附效果的影响。结果表明,上样流速在1~4 mL/min范围内,3种雌激素的色谱峰面积随上样流速的增加一直呈下降趋势,但过慢的上样流速势必会影响到整个富集分析过程的效率。综合考虑,最终选择2 mL/min作为适宜的上样流速。
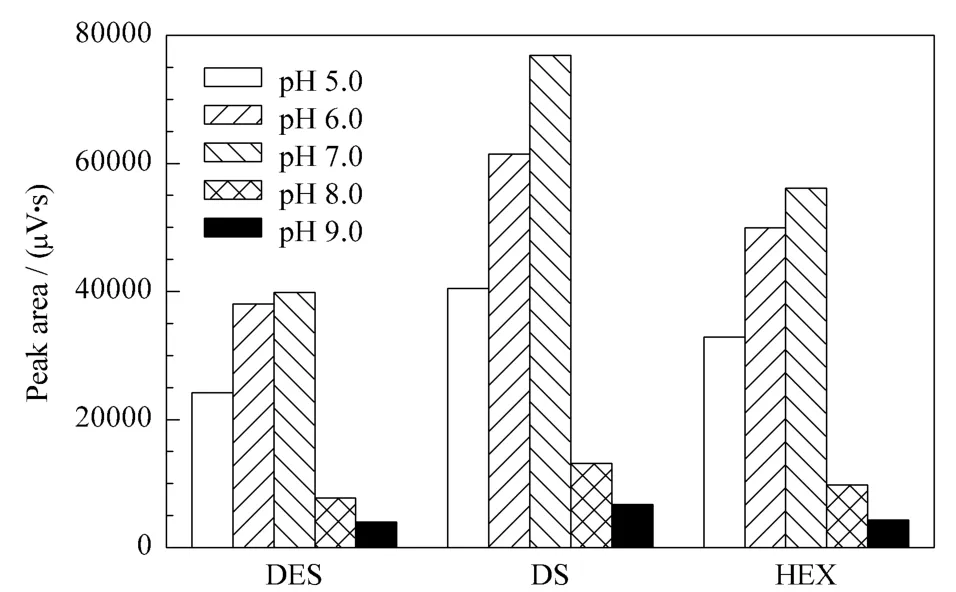
图2 样品溶液pH对雌激素富集效果的影响Fig.2 Effect of pH of the sample solution on extraction efficiencies of the estrogens
2.3 方法学考察
2.3.1 直接进样测定的标准曲线
将3种分析物的混合标准液稀释成质量浓度为0.5、1、2、5、8、10 mg/L的系列标准溶液,依次进样分析,每个浓度样品平行测定3次,对分析物的峰面积(Y,μV·s)与质量浓度(X,mg/L)进行线性回归,绘制标准曲线。3种雌激素的线性方程见表1。
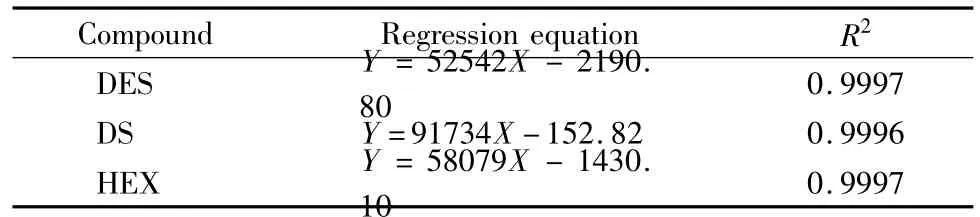
表1 3种雌激素的线性方程与相关系数(R2)Table 1 Regression equations and correlation coefficients(R2)of the three estrogens
2.3.2 方法的线性关系、精密度和检出限
为了考察方法的线性关系,配制质量浓度分别为0.5、1、2、5、8、10 μg/L 的 3 种雌激素混合标准工作液,在线富集100 mL,每个浓度样品平行测定3次,对分析物的峰面积(Y,μV·s)与质量浓度(X,μg/L)进行线性回归,所得线性方程即为富集标准曲线方程(见表2)。以5 μg/L的混合标准工作液富集100 mL,重复测定5次,计算各待测组分的平均峰面积,计算方法的相对标准偏差(RSD),其结果见表3。由表2和3可以看出,各待测组分的R2>0.99,同时各待测组分峰面积的RSD<7%,说明该方法的线性关系良好,且适用于雌激素残留的痕量检测与定量分析。根据S/N=3求得3种雌激素的检出限(LOD),其结果见表3。
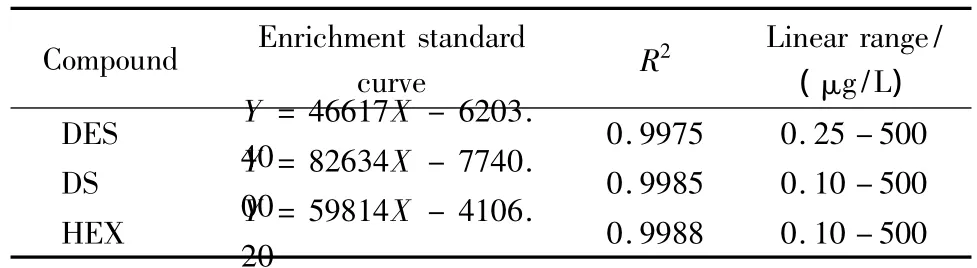
表2 3种雌激素的富集标准曲线方程、相关系数及线性范围Table 2 Enrichment standard curves,correlation coefficients and linear ranges of the three estrogens
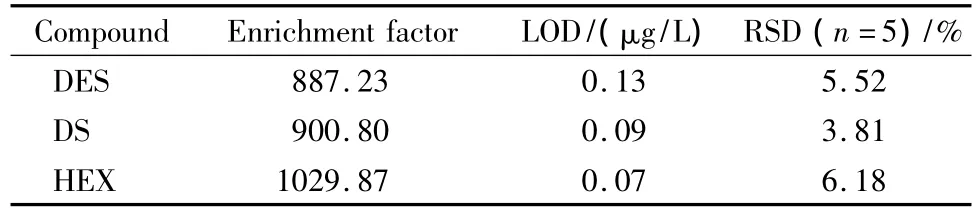
表3 3种雌激素的富集倍数、方法的检出限及相对标准偏差Table 3 Enrichment factors,LODs and RSDs of the three estrogens
2.3.3 富集倍数
合成材料对于3种待测雌激素的富集倍数即为富集标准曲线与直接进样标准曲线斜率的比值[16],其结果见表3。富集倍数(R)的计算公式为:R=K1/K2×1000,其中:K1为富集标准曲线的斜率,K2为直接进样标准曲线的斜率。
2.4 实际样品的测定
为了评价方法的适用性,以矿泉水和自来水作为样品测定雌激素的残留。经检测,所选择的样品中均未检出雌激素残留。为了进一步验证所选择样品前处理方法的可行性,对样品进行添加回收试验。采用3个添加水平,每个水平重复测定3次,计算添加回收率,结果见表4。可以看出,矿泉水和自来水中3种雌激素的添加回收率范围分别为83.46%~99.43%与82.31%~98.54%,且RSD<7.2%,可满足水样中雌激素残留的痕量检测需要。从图3可以看出,样品经前处理富集,残留杂质的出峰时间在6 min之前,对于待测目标物的检测没有影响。
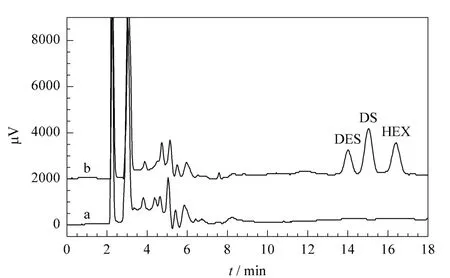
图3 (a)空白矿泉水样品和(b)加标(0.5 μg/L)矿泉水样品的色谱图Fig.3 Chromatograms of(a)a blank sample and(b)the sample spiked with the estrogens at 0.5 μg/L
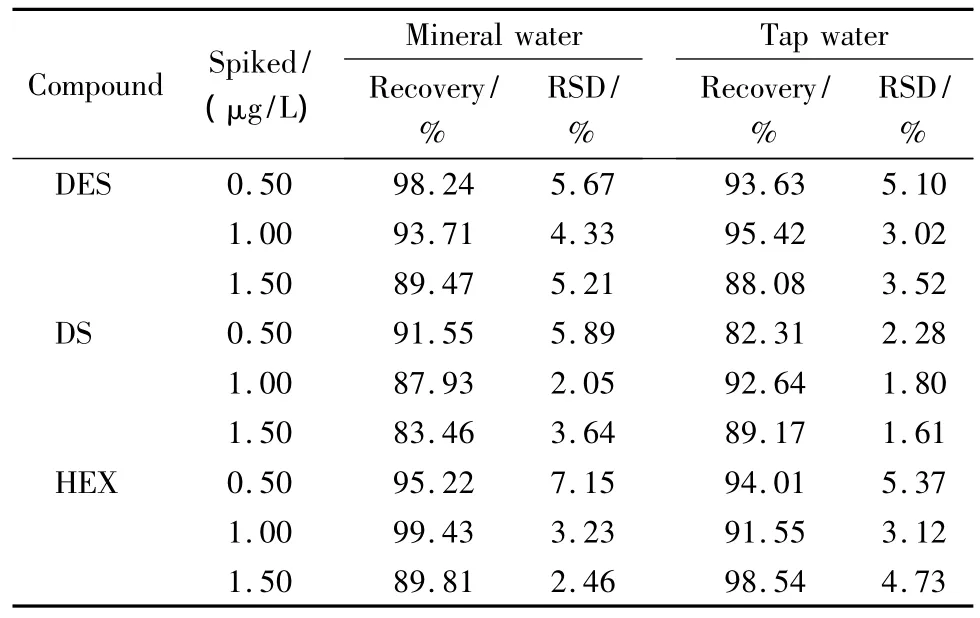
表4 不同加标水平下雌激素的回收率(n=3)Table 4 Recoveries of the estrogens spiked at three levels in the samples(n=3)
3 结论
本研究以溶胶凝胶技术合成的聚合物为固相萃取填充材料,建立了固相萃取与高效液相色谱联用在线富集测定水样中3种雌激素残留的痕量检测方法,优化了影响富集效果的若干因素。结果表明,在优化的条件下,3种雌激素的检出限、加标回收率及RSD均可满足定量分析的需要。该方法简便、高效、准确,适用于此类雌激素的残留监控。
[1]Ren Q L,Ma Q,Bai H J,et al.Journal of Henan Agricultural Sciences(任巧玲,马强,白红杰,等.河南农业科学),2006(4):114
[2]Janowski T,Zdunczyk S,Malecki-Tepicht J,et al.Domest Anim Endocrin,2002,23(1/2):125
[3]Ganmaa D,Sato A.Med Hypotheses,2005,65:1028
[4]Farlow D W,Xu X,Veenstra T D,et al.J Chromatogr B,2009,877(13):1327
[5]Newmark H L,Heaney R P.Nutr Cancer,2010,62(3):297
[6]Parodi P W.Int Dairy J,2012,22(1):3
[7]Tang X S,Zhang Q X,Du X F,et al.Science and Technology of Food Industry(唐晓姝,张秋香,杜先锋,等.食品工业科技),2013,34(6):53
[8]Lü Y K,Yan X P.Chinese Journal of Analytical Chemistry(吕运开,严秀平.分析化学),2005,33(2):254
[9]Jiang X,Zhao C,Jiang N,et al.Food Chem,2008,108(3):1061
[10]Sadowski R,Gadzala-Kopciuch R.J Sep Sci,2013,36:2299
[11]Zhang Q,Liu Y.Modern Food Science and Technology(张群,刘烨.现代食品科学),2009,25(3):337
[12]Li X,Mou G Q,Chen L J,et al.Chinese Journal of Chromatography(李雪,牟光庆,陈历俊,等.色谱),2013,31(9):908
[13]Deng X L,Fan J,Huang T H,et al.China Food(邓晓丽,范军,黄涛宏,等.中国食品),2011(4):56
[14]Zhu Y L,Shao D S.Chinese Journal of Veterinary Drug(朱永林,邵德胜.中国兽药杂志),2006,40(11):5
[15]He J X,Fang G Z,Wang S.J Chromatogr A,2006,1127:12
[16]He J X,Wang S,Fang G Z.J Agric Food Chem,2008,56:2919
