高效液相色谱法检测新西兰Manuka蜂蜜中的甲基乙二醛
2014-10-22费晓庆沈崇钰
陈 磊,栾 军,费晓庆,吴 斌,沈崇钰,张 睿
(江苏出入境检验检疫局动植物与食品检测中心,江苏 南京 210001)
Manuka蜂蜜是新西兰特有的一种珍贵蜜种,是蜜蜂采集新西兰特有的一种红茶树——Manuka(Leptospermum scoparium)的花蜜酿造而成的。它区别于其他蜂蜜之处在于Manuka蜂蜜有强大而独特的抗菌活性。蜂蜜一般都具有抗菌活性,这是由蜂蜜本身的高渗透性、较强的酸度并且一般含有过氧化物造成的[1,2]。而 Manuka蜂蜜的抗菌活性则不依赖于过氧化物,因此被称为非过氧化抗菌活性(non-peroxide antibacterial activity,NPA)。1991年,Molan等[3]最早报道了这种抗菌活性的物质基础,并将其命名为Manuka独有因子(unique Manuka factor,UMF);由于并不清楚UMF的准确分子结构,采用微生物学方法对Manuka蜂蜜的NPA进行了检测。通过比较Manuka蜂蜜和一系列不同浓度的苯酚溶液对金黄色葡萄球菌的抑制程度,从而对Manuka蜂蜜的NPA进行评级,并标示为UMF5+、UMF10+、UMF15+等等(数字表示苯酚溶液的浓度)。目前,UMF已经被新西兰UMF蜂蜜协会(U-nique Manuka Factor Honey Association,UMFHA)作为评价Manuka蜂蜜的质量指标,并要求被授权的蜂蜜生产商将其标注在产品包装上。
2008 年,Mavric等[4]发现甲基乙二醛(methylglyoxal,MGO)可能是Manuka蜂蜜具有NPA的主要物质基础。同年,Adams等[5]也报道了相同的结论,并且发现Manuka蜂蜜中MGO的含量与NPA的相关性高达0.98。由于采用微生物方法检测NPA操作比较繁琐,结果变异较大,而且物质基础不明确,因此部分研究人员呼吁采用MGO的含量来代替UMF值用于标示Manuka蜂蜜的NPA。但是,也有部分研究人员认为还没有充分的证据证明MGO能完全代表Manuka蜂蜜的NPA,而且UMF更能体现新西兰Manuka植物的地域独特性和绿色环保理念。因此,目前市场上销售的Manuka蜂蜜的产品包装上会有UMF和MGO两种指标来标示Manuka蜂蜜的抗菌活性。
目前国内对Manuka蜂蜜的研究较少,且主要集中在其抗菌活性方面。朱威等[1]和玄红专等[2]综述了国外对Manuka蜂蜜抗菌活性的研究。孙艳萍等[6]研究了 Manuka UMF25+、UMF10+ 以及其他7种蜂蜜对铜绿假单胞菌的体外抗菌活性,结论为9种蜂蜜在体外均可抑制铜绿假单胞菌,进口蜜更佳。对Manuka蜂蜜中相关物质的检测方法,仅见吕辰等[7]采用高效液相色谱-串联质谱法对多种蜂蜜中的5种双稠吡咯啶类生物碱进行了筛查,结果发现野百合碱、克氏千里光宁和倒千里光碱均未检出,而千里光菲啉和千里光宁在大多数蜂蜜中均能检出。
MGO属于二羰基化合物,国内报道的检测方法集中在大气环境监测领域。李阳等[8]建立了一种五氟化苯肼衍生化-热解吸-GC/MS方法对大气中23种羰基化合物进行了测定。牟翠翠等[9]和冯艳丽等[10]分别报道了2,4-二硝基苯肼衍生-高效液相色谱法用于测定大气中二羰基化合物。邹婷等[11]采用五氟苄基羟胺衍生与GC/MS联用的方法分析了大气中的单羰基化合物和多羰基化合物。对于蜂蜜中MGO的检测方法,目前国内尚无相关文献报道。对此,本实验室开发了高效液相色谱法对Manuka蜂蜜的MGO含量进行检测。
1 实验部分
1.1 仪器与试剂
Agilent 1200液相色谱仪,配有自动脱气机、二元高压泵、自动进样器、柱温箱、二极管阵列检测器及ChemStation数据处理软件(美国Agilent公司);PURELAB Option-Q15纯水仪(英国ELGA公司);电子天平(精确到0.00001 g,德国Sartorius公司;精确到0.01 g,瑞士METTLER TOLEDO公司)。
MGO对照品(40%水溶液)购自美国Sigma-Aldrich公司;邻苯二胺(o-phenylenediamine,OPD)购自阿拉丁化学试剂(上海)有限公司;甲醇(色谱纯,德国 Merck公司);水为超纯水(18.2 MΩ·cm);0.22 μm 有机滤膜(德国 Membrana公司)。
Manuka蜂蜜样品由新西兰UMF蜂蜜协会提供,中国蜂蜜样品由通标标准技术服务有限公司提供。
1.2 标准溶液的配制
精密量取MGO对照品100 μL,用水稀释并定容至10 mL,配成4 g/L的标准储备液(4℃避光保存)。临用前用水将标准储备液逐级稀释为适当浓度的标准工作溶液。
精密称取邻苯二胺0.6 g,用水溶解并定容至100 mL,配成6 g/L的邻苯二胺水溶液。
1.3 样品前处理
称取1 g蜂蜜溶于10 mL水中,取1 mL该蜂蜜水溶液与1 mL邻苯二胺水溶液(6 g/L)混匀,在室温、避光条件下进行衍生化反应8 h以上,过0.22 μm滤膜后进样分析。
1.4 液相色谱条件
色谱柱:Kromasil反相色谱柱(150 mm×4.6 mm,5 μm);流动相:0.1%(v/v)乙酸水溶液(A)和甲醇(B);检测波长:318 nm;流速:1.0 mL/min;柱温:30℃;进样量:10 μL;保留时间:10.3 min。梯度洗脱程序:0~5 min,30%B;5~10 min,30%B~90%B;10~15 min,90%B;15~16 min,90%B~30%B;16~20 min,30%B。
2 结果与讨论
2.1 衍生化反应条件的优化
MGO的紫外吸收弱,需衍生化反应后再进行检测。MGO 属于 1,2-二羰基化合物,Weigel等[12]报道1,2-二羰基化合物能与邻苯二胺反应生成喹噁啉类化合物(见图1),在反相色谱柱上有较好的保留,且紫外吸收强。Mavric等[4]和 Oelschlaegel等[13]建立的MGO检测方法中的衍生化条件均是在Weigel等[12]报道的基础上进行了修改,均为蜂蜜水溶液与邻苯二胺水溶液在室温、避光条件下反应8 h以上,只是蜂蜜样品和邻苯二胺溶液的浓度不同。本实验在室温、避光、反应时间(8 h以上)等衍生化条件不变的基础上,对衍生化试剂的浓度进行了优化。
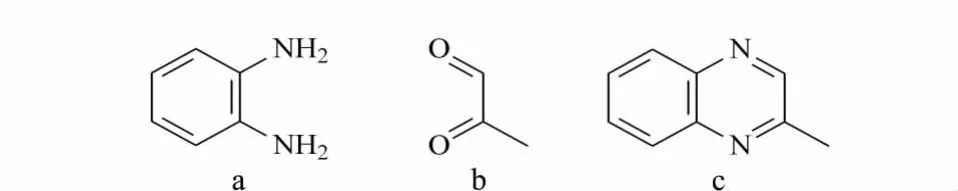
图1 (a)邻苯二胺、(b)MGO和(c)MGO衍生化产物的化学结构式Fig.1 Chemical structures of(a)OPD,(b)MGO and(c)MGO derivative
将100 mg/L的MGO标准溶液分别与1、2、4、6、8、10 g/L的邻苯二胺水溶液按体积比1∶1混合,进行衍生化反应。通过比较MGO衍生化产物的峰面积考察衍生化效率,结果如图2所示。
从图2中可以看出,邻苯二胺水溶液的浓度从1 g/L升高到6 g/L,衍生化产物的量逐渐上升;从6 g/L升高到10 g/L,衍生化产物的量开始趋于稳定,提示反应到达完全。因此,本实验选择6 g/L的邻苯二胺水溶液。
此外,由于蜂蜜直接稀释后的水溶液含糖量高,对色谱柱伤害较大,本实验对衍生化反应中蜂蜜水溶液的浓度也进行了调整。Mavric等[4]和 Weigel等[12]报道的方法中蜂蜜水溶液的浓度分别为150和300 g/L,本实验中蜂蜜水溶液的浓度为100 g/L。并且与邻苯二胺溶液等体积混合,因此用于进样的溶液中只含有50 g/L的蜂蜜,能尽量避免高糖基质对色谱柱的损伤。在本实验的方法学验证和实际样品检测完成前后,色谱柱压力和MGO衍生化产物的保留时间均未见明显改变。
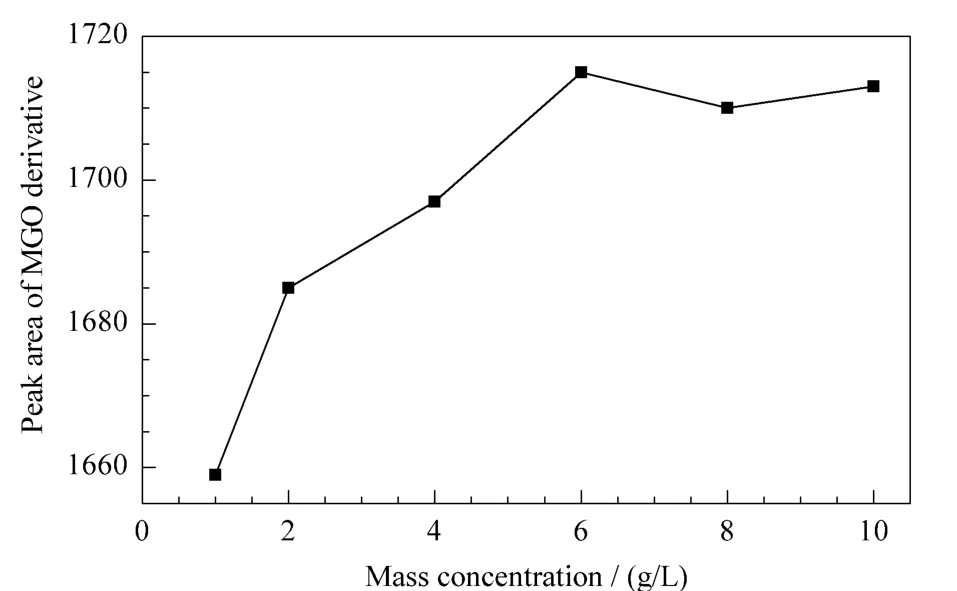
图2 邻苯二胺浓度对衍生化效率的影响Fig.2 Effect of the mass concentration of OPD on the efficiency of derivatization
2.2 检测波长的选择
文献报道的 MGO 检测波长为 312 nm[4,12]或316 nm[13]。本实验中,通过光谱扫描发现,最佳的检测波长为318 nm。
2.3 线性范围、检出限和定量限
MGO标准溶液与邻苯二胺水溶液进行衍生化反应后进行检测,以衍生化产物的峰面积Y对MGO的质量浓度X(mg/L)进行线性回归,在1~50 mg/L范围内线性良好,相关系数为0.9999。检出限(S/N=3)为 0.02 mg/L,定量限(S/N=10)为 0.06 mg/L。HPLC图谱见图3,在MGO衍生化产物的保留时间附近并没有干扰峰存在。
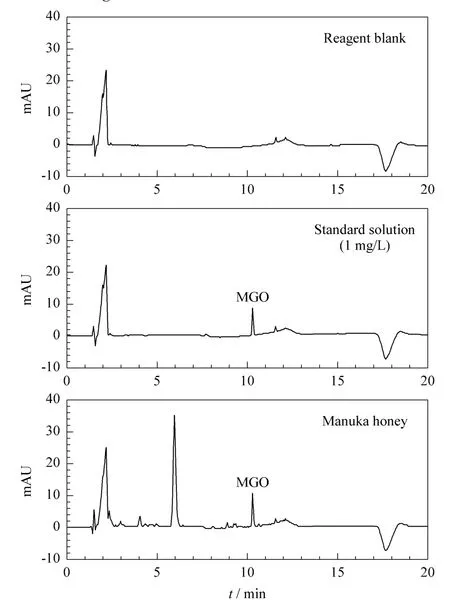
图3 试剂空白(水)及MGO标准溶液(1 mg/L)和Manuka蜂蜜样品的衍生产物的HPLC图谱Fig.3 HPLC chromatograms of reagent blank(water)and the derivatives of MGO standard solution(1 mg/L)and Manuka honey
Mavric 等[4]、Weigel等[12]和 Oelschlaegel等[13]报道的MGO检出限分别为0.2、0.2、5 mg/kg,本方法的检出限为0.02 mg/L,即0.4 mg/kg;差异的原因可能是所使用的蜂蜜水溶液浓度不同。Mavric等[4]和 Weigel等[12]报道的检出限较低,但蜂蜜水溶液浓度较高(150和30 g/L),高糖分对色谱柱损伤较大;Oelschlaegel等[13]使用的蜂蜜水溶液浓度很低(15 g/L),但检出限太高。本实验条件中蜂蜜水溶液浓度介于两者之间,在保证灵敏度的前提下,能尽量减少高糖基质对色谱柱的损伤。而且,Manuka蜂蜜样品中MGO的含量一般较高,本实验的检出限能够满足实际样品的检测需要。
2.4 精密度和回收率
在Manuka蜂蜜样品中分别添加50.0、100.0、200.0 mg/kg的 MGO,每个添加水平重复 5次,HPLC检测后计算精密度和回收率。3个添加水平下的RSD分别为1.26%、0.89%和0.69%,平均回收率分别为98.3%、101.5%和99.5%(见表1)。结果表明,蜂蜜中的其他物质对MGO衍生化产物的检测准确度并没有影响。

表1 Manuka蜂蜜中MGO的精密度和回收率(n=5)Table 1 Precision and recovery of MGO spiked in Manuka honey(n=5)
2.5 衍生化产物的稳定性
在 Manuka蜂蜜样品中分别添加50、100、200 mg/kg的MGO,每个添加水平重复3次,在衍生化反应结束后0 h和24 h分别检测,考察衍生化产物的稳定性。结果如图4所示,衍生化产物在室温放置24 h后仍稳定,蜂蜜中的其他物质对MGO衍生化产物的稳定性没有影响。
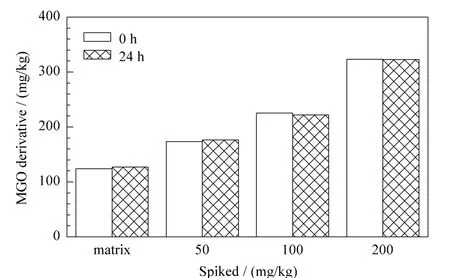
图4 MGO衍生化产物的稳定性(n=3)Fig.4 Stability of MGO derivative(n=3)
2.6 蜂蜜样品的检测
利用本方法对61个Manuka蜂蜜样品中的MGO含量进行了检测。其中,UMF标示值为5+的蜂蜜样品共14个,MGO含量为(188.0±62.1)mg/kg;UMF标示值为10+的蜂蜜样品共16个,MGO含量为(324.6±90.4)mg/kg;UMF标示值为15+的蜂蜜样品共12个,MGO含量为(523.7±59.1)mg/kg;UMF标示值为20+的蜂蜜样品共13个,MGO含量为(726.1±76.9)mg/kg;UMF标示值为 25+的蜂蜜样品共 6个,MGO含量为(1035.0±75.8)mg/kg。该检测结果与 Adams等[5]和 Stephens等[14]的报道基本相符。
此外,利用本方法对中国不同产地的油菜蜜、荆条蜜、洋槐蜜、椴树蜜、葵花蜜、枣花蜜、荞麦蜜、棉花蜜共16个样品中的MGO含量进行了检测。结果显示,MGO 含量均≤1 mg/kg。Stephens等[14]报道,新西兰Kanuka蜜、三叶草蜜、Rewarewa蜜和甘露蜜中MGO的含量均远低于 Manuka蜜。Arena等[15]报道,意大利刺槐蜜、栗子蜜、柑橘蜜、桉树蜜、甘露蜜以及多花种混合蜂蜜中MGO的含量均≤3 mg/kg。以上研究结果均显示,MGO为Manuka蜂蜜中的特有成分。本文建立的MGO含量检测方法,可以为我国进口Manuka蜂蜜的质量评价提供参考。
3 结论
建立了高效液相色谱法用于检测新西兰Manuka蜂蜜中的甲基乙二醛。该方法前处理简单,具有良好的灵敏度、回收率和重复性,可用于新西兰进口蜂蜜的质量控制,也适用于中国蜂蜜中甲基乙二醛的检测。
[1]Zhu W,Hu F L,Li Y H,et al.Natural Product Research and Development(朱威,胡福良,李英华,等.天然产物研究与开发),2004,16(4):372
[2]Xuan H Z,Hu F L.Journal of Bee(玄红专,胡福良.蜜蜂杂志),2002(9):23
[3]Allen K L,Molan P C,Reid G M.J Pharm Pharmacol,1991,43(12):817
[4]Mavric E,Wittmann S,Barth G,et al.Mol Nutr Food Res,2008,52(4):483
[5]Adams C J,Boult C H,Deadman B J,et al.Carbohydr Res,2008,343(4):651
[6]Sun Y P,Li P.Occupation and Health(孙艳萍,李萍.职业与健康),2012,28(19):2366
[7]Lü C,Ding T,Ma X,et al.Chinese Journal of Chromatography(吕辰,丁涛,马昕,等.色谱),2013,31(11):1046
[8]Li Y,Shao M,Lu S H.Environmental Chemistry(李阳,邵敏,陆思华.环境化学),2009,28(5):630
[9]Mu C C,Feng Y L,Zhai J Q,et al.Chinese Journal of Analytical Chemistry(牟翠翠,冯艳丽,翟金清,等.分析化学),2010,38(11):1573
[10]Feng Y L,Mu C C,Fu Z R,et al.Chinese Journal of Analytical Chemistry(冯艳丽,牟翠翠,付正茹,等.分析化学),2011,39(11):1653
[11]Zou T,Feng Y L,Fu Z R,et al.Acta Scientiae Circumstantiae(邹婷,冯艳丽,付正茹,等.环境科学学报),2012,32(11):2718
[12]Weigel K,Opitz T,Henle T.Eur Food Res Technol,2004,218(2):147
[13]Oelschlaegel S,Gruner M,Wang P N,et al.J Agric Food Chem,2012,60(29):7229
[14]Stephens J M,Schlothauer R C,Morris B D,et al.Food Chem,2010,120(1):78
[15]Arena E,Ballistreri G,Tomaselli F,et al.J Food Sci,2011,76(8):C1203
