在线凝胶渗透色谱-二维气相色谱/质谱法测定鲫鱼样品中的14种农药残留
2014-10-22李淑静宓捷波陈其勇葛宝坤翟自芹
李淑静,董 梅,许 泓,宓捷波,陈其勇,葛宝坤,翟自芹
(1.天津出入境检验检疫局动植物与食品检测中心,天津 300461;2.天津大学材料科学与工程学院,天津 300072)
与植物源性食品不同,动物源性食品具有基质复杂、多样等特点,通常会对分析结果产生干扰,需通过样品前处理(如提取、净化等)降低基质效应。目前发展了多种提取和净化方式,如冷冻离心、液-液分离、凝胶渗透色谱(GPC)、固相萃取(SPE)和固相微萃取(SPME)。其中,GPC对于从高相对分子质量物质(Mr=600~1500,如多肽、脂肪)中分离低相对分子质量的物质(Mr<400,如农药)具有非常好的效果[1],因此GPC常用于高脂肪样品的目标物净化[2]。作为一种非常有效的前处理方式,GPC根据体积排阻的原理将不同相对分子质量的物质进行分离,能很好地去除样品基质中可能干扰目标化合物的油脂、色素、生物碱、聚合物等高分子化合物[3]。GPC作为一种样品净化手段,在国内外应用的已经比较普遍,尤其在富含脂肪、色素等大分子的样品分离净化方面,GPC具有明显的优势。使用GPC可以提高仪器的利用效率、延长分析柱寿命、提高分析精确度和准确度[4-6]。GPC的使用方式主要有离线和在线两种。离线前处理方法往往耗时、成本高并且重现性差,而在线GPC可以降低分析时间和成本。研究表明在线GPC-GC/MS配合程序升温气化(PTV)进样模式适用于多残留分析[7]。
食品中农药残留的检测手段主要以气相色谱[8]、气相色谱-质谱联用技术[9,10]为主。欧阳运富等[11]利用气相色谱-质谱联用技术测定蔬菜、水果中的多农药残留,定量限为1 ~6 μg/kg;Yang等[12]采用配备双脉冲火焰光度检测器的气相色谱测定动物源性食品中的49种有机磷农药残留;Wu等[13]利用加速溶剂萃取-气相色谱/质谱法测定动物源性食品中109种农药残留,检出限可达0.3 μg/kg。但是由于动物源性食品具有基质多样和复杂的特点,基质往往会对检测结果产生干扰[7]。因此需要通过复杂的提取、净化步骤降低基质对检测结果的干扰,而复杂的前处理过程要耗费大量的有机溶剂,既增加了分析成本又污染了环境。目前新发展起来的二维气相色谱(MDGC)由于峰容量和分离效率大大提高,成为应用于多残留分析的强有力分析手段。MDGC的工作原理是将两根色谱柱串接,通过中心切割的方式,将目标物选择性切割进入二维色谱柱进行分离。MDGC已经用于尿液中的双酚A和双酚 B 的检测[14];Castillo 等[15]采用固相微萃取-MDGC-MS测定果汁中的46种农药残留。将在线GPC与MDGC/MS技术联用可以同时借助两项技术的优势,实现多类农药的同时分离分析,而目前国内外并没有相关的文献报道。
基于此,本研究采用在线GPC-MDGC/MS技术测定动物源性食品中的多类农药残留。分别选取了3大农药类型中比较常见且典型的14种农药作为研究对象(14种农药为有机磷农药中的灭线磷、甲基对硫磷、杀螟硫磷、氯唑磷、乐果;有机氯农药中的δ-六六六、六氯苯、五氯硝基苯、林丹、五氯苯胺和菊酯类农药七氟菊酯、苄呋菊酯、甲氰菊酯、苯醚菊酯)。样品经净化后直接由系统导入MDGC/MS进行分析。经GPC净化后的样品去除了对仪器和分析结果具有损害和干扰影响的物质,如脂肪和大分子蛋白质,为样品分析提供了良好的内部环境,再加上配有PTV大体积进样口,最大进样量可达20 μL,故在线GPC-MDGC/MS分析系统的灵敏度和稳定性均优于一般的GC-MS系统。
1 实验部分
1.1 仪器及试剂
在线GPC-MDGC/MS装置配有电子轰击(EI)离子源(岛津QP2010 Plus);高速均质器(IKA T25 digital,Turrax,德国);离心机(Sigma 3-18K,德国);Milli-Q纯水仪(Millipore公司);0.22 μm滤膜;旋转蒸发器(BUCHI ROTAVAPOR R-215,瑞士);伯仲胺(PSA)粉末(100 g,Agilent Technology)。
丙酮、环己烷、乙酸乙酯(色谱纯);中性氧化铝(100~200目);14种农药标准样品(纯度>95%):灭线磷、δ-六六六、甲基对硫磷、杀螟硫磷、六氯苯、五氯硝基苯、林丹、乐果、氯唑磷、五氯苯胺、七氟菊酯、苄呋菊酯、甲氰菊酯、苯醚菊酯;内标物为环氧七氯(Dr.Ehrenstorfer公司)。
1.2 标准储备液的配制
准确称取14种农药标准品和内标物各0.01 g,置于10 mL棕色容量瓶中,用丙酮定容,配制成1 g/L的标准储备液,置于4℃冰箱中保存。
1.3 样品提取
称取5 g试样(精确至0.01 g),放入盛有10 g无水硫酸钠和2 g中性氧化铝的50 mL的离心管中,加入18 mL环己烷/乙酸乙酯(1∶1,v/v)混合溶剂,均质提取1.5 min,于3000 r/min下离心3 min。将上清液过装有无水硫酸钠的筒形漏斗,收集于100 mL鸡心瓶中;残渣用18 mL的环己烷/乙酸乙酯(1∶1,v/v)混合溶剂重复提取1次,经离心过滤后,合并2次提取液;将提取液于40℃水浴旋转蒸发器中旋转蒸发至约2 mL。将上述浓缩液转移至10 mL离心管中,用5 mL环己烷/乙酸乙酯(1∶1,v/v)混合溶剂分两次洗涤鸡心瓶并转移至上述离心管中,氮吹至近干。用丙酮/环己烷(3∶7,v/v)定容至2 mL,加入100 mg的PSA粉末,涡旋1 min,放入离心机中在5000 r/min下离心5 min;然后放入-18℃的冰箱中冷冻2 h。冷冻后用0.22 μm滤膜将样液过滤入样品瓶中待净化、检测。
1.4 GPC分析条件
GPC色谱柱:Shim-pack VP-ODS(150 mm×4.6 mm,5 μm);流动相:丙酮/环己烷(30∶70,v/v);泵A和B流量:0.10 mL/min;光电二极管阵列检测器(PDA)检测波长:254 nm;柱温:40℃;进样量:10 μL;馏分切割时间:3.70~5.70 min;具体馏分切割程序见表1。

表1 GPC馏分切割程序Table 1 Function cutting program of GPC
1.5 MDGC分析条件
一维条件 色谱柱:DB-5MS毛细管柱(15 m×0.25 mm×0.1 μm)。PTV进样口;不分流恒压模式。升温程序:90℃保持5 min,以80℃/min升温至290℃,保持30 min。柱温箱升温程序:60℃保持5 min,以8℃/min升温至90℃,保持1 min;再以20℃/min升温至230℃;最后以15℃/min升温至290℃,保持10 min。载气:高纯氦气;总流量:30 mL/min;柱流量:3.23 mL/min;线速度:34.4 cm/s;定量环体积:200 μL。
二维条件 色谱柱:DB-17MS毛细管柱(30 m×0.25 mm×0.25 μm)。柱温箱升温程序:60℃保持18 min,以10℃/min升温至80℃,保持5 min;再以15℃/min升温至230℃,保持3 min;最后以25℃/min升温至290℃,保持7 min。载气:高纯氦气;柱头压力:120 kPa。
一维检测器:氢火焰离子化检测器(FID);温度:300℃;H2流量:40.0 mL/min;空气流量:400 mL/min;尾吹气(He)流量:30.0 mL/min。
二维检测器:质谱检测器,EI源,离子源温度200℃;接口温度:260℃;溶剂切割时间:15 min;扫描时间:16~48 min;扫描模式:选择离子扫描(SIM);14种农药的定性离子、定量离子和保留时间见表2。
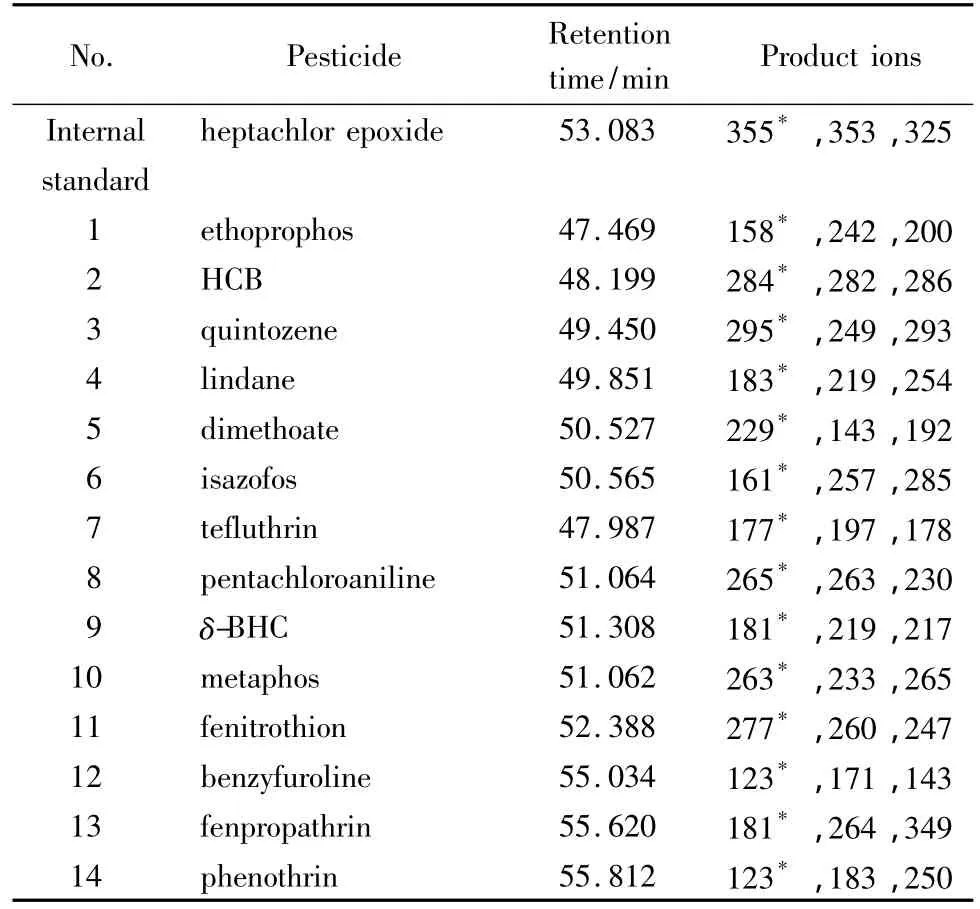
表2 14种农药的保留时间和定量、定性离子Table 2 Retention times,quantitative ions and qualitative ions of the 14 pesticides
2 结果与讨论
2.1 MDGC的工作流程
本研究的整个工作流程见图1。首先待测样品经过在线GPC的净化,之后含有待测组分的馏分进入定量环,然后通过阀的切换进入气相色谱系统。一维色谱采用FID作为检测器,对各组分在第一维色谱柱上的保留时间进行监测。根据一维保留时间通过中心切割的方式将感兴趣或未分离组分送入第二维色谱柱进行分离,最后由质谱给出定性和定量结果。
2.2 在线GPC条件的确定
MDGC通过排空阀排出GPC洗脱时间短的大分子油脂和色素(叶绿素、叶黄素)、生物碱、聚合物等大分子化合物,将目标化合物导入捕集环路,然后经MDGC分离最后进入质谱进行检测。如果切入的起始时间过早或者切入的结束时间过迟,油脂、色素等成分的去除效果会降低;如果切入的时间过迟或者结束的时间过早,目标化合物就不能全部切入气相色谱系统;所以必须根据实际样品的情况来选择最佳的切割时间。因此,根据目标化合物中相对分子质量最小的灭线磷(Mr=242)和相对分子质量最大的七氟菊酯(Mr=418)在GPC柱上的保留时间,确定GPC流出液的收集时间为3.70~5.70 min。
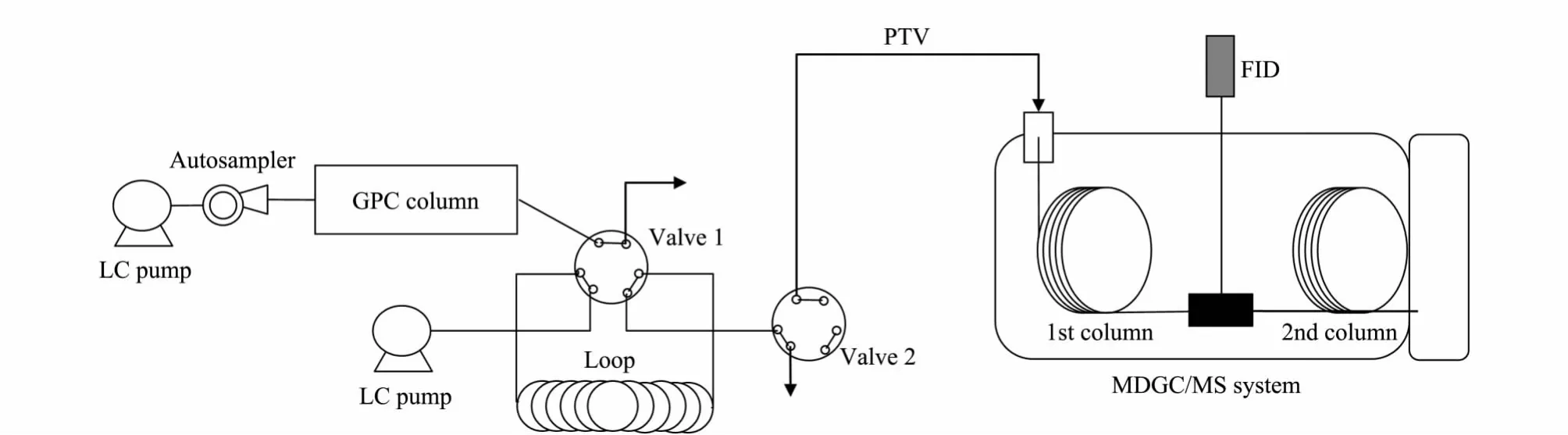
图1 在线GPC-MDGC/MS工作流程示意图Fig.1 Schematic diagram of on-line GPC-MDGC/MS
应用在线GPC系统可在50 min内完成从GPC提纯分析到检测的过程。溶剂消耗只有10 mL,是常规样品前处理的几十分之一,大大减少了有机溶剂的消耗量,不仅能降低费用,而且有助于减少有机溶剂对环境的危害。
2.3 二维切割时间的确定及分离效率
一维色谱采用FID作为检测器,由于FID针对不同目标化合物响应值有差别,目标化合物的一维分离为预分离阶段,可以实现目标化合物的组分离(见图2a)。从该图中可以看出,14种农药在FID上响应值较低,且分离效果较差,但是可以将14种化合物分4次进行切割,即:18~20 min、21.2~25.2 min、30.7 ~31.25 min、31.75 ~34 min,通过 4次切割可将目标化合物分4次送入二维色谱进行进一步的分离分析。
图2b为经过二维分离后由质谱检测器获得的色谱图。从该图中可以看出,在一维色谱中无法分离的组分经过二维分离后可以得到有效的分离。因此,通过二维切割的方式可以将一维色谱未分离组分送入二维色谱进一步分离,从而达到彻底分离的目的。
2.4 监测离子对的选择
配制14种农药混合标准溶液(1 mg/L),按照仪器条件进行全扫描。根据各农药的全扫描质谱图及背景干扰情况,对定量、定性离子进行选择,尽量选择质荷比和丰度较大且基质在被测组分保留时间干扰最小的特征离子作为定量、定性离子,结果见表2。图2b为14种农药标准物质的选择离子监测色谱图。
2.5 方法的线性范围和相关系数
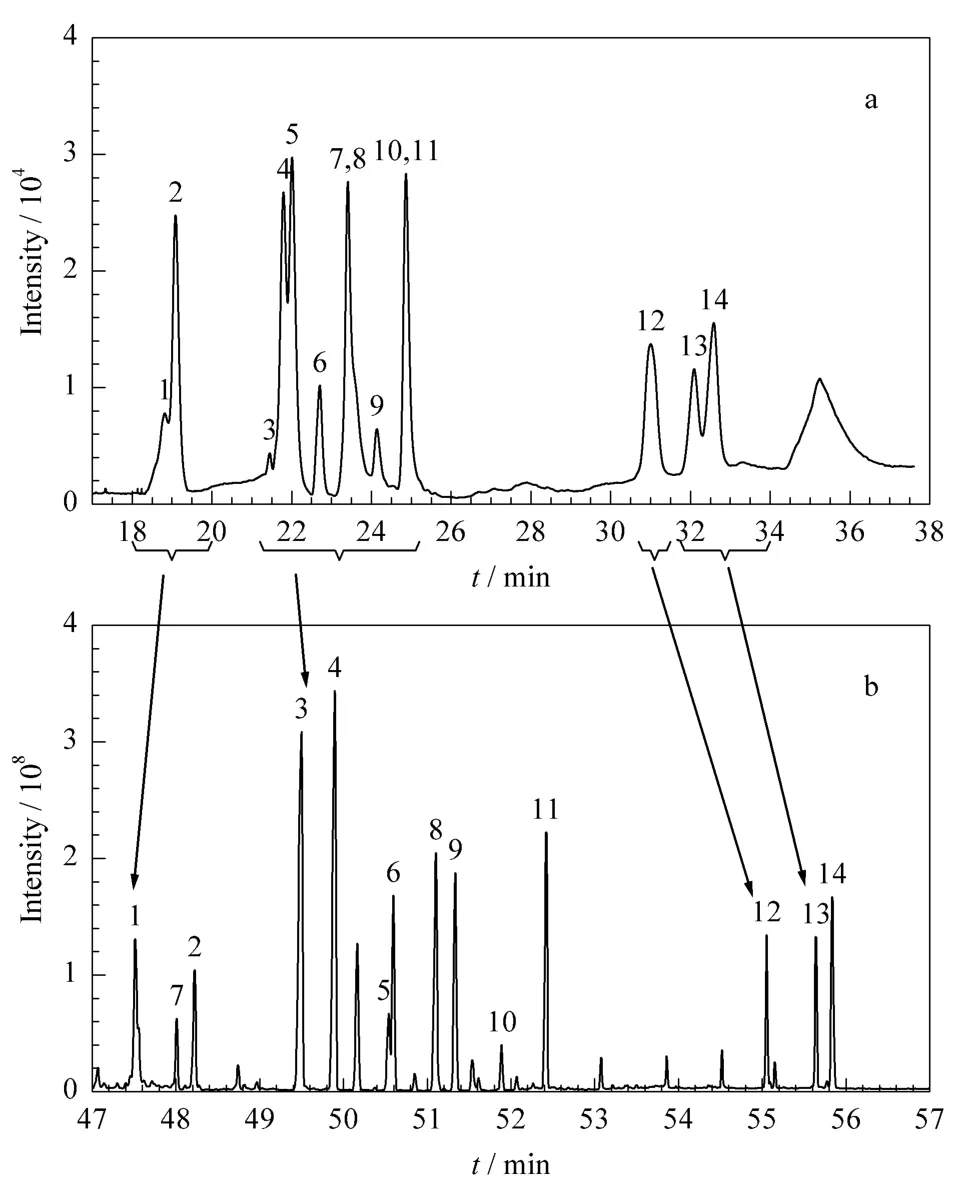
图2 14种农药的(a)一维色谱图和(b)二维色谱图Fig.2 (a)First dimensional chromatogram and(b)second dimensional chromatogram of the 14 pesticides
用丙酮将标准储备液逐级稀释到质量浓度分别为0、0.025、0.125、0.25、0.75 mg/L,内标物质量浓度为0.2 mg/L,按照优化的分析条件进行线性分析,每个样品重复测定6次,取平均值。以目标物和内标物峰面积之比为纵坐标(Y),以目标物和内标物质量浓度之比为横坐标(X)绘制工作曲线。结果表明,14种农药的质量浓度在0.025~0.75 mg/L范围内与其峰面积呈良好的线性相关性,相关系数(r2)>0.99(见表3)。根据10倍S/N和目标化合物的限量要求制定方法的定量限为0.01 mg/kg。
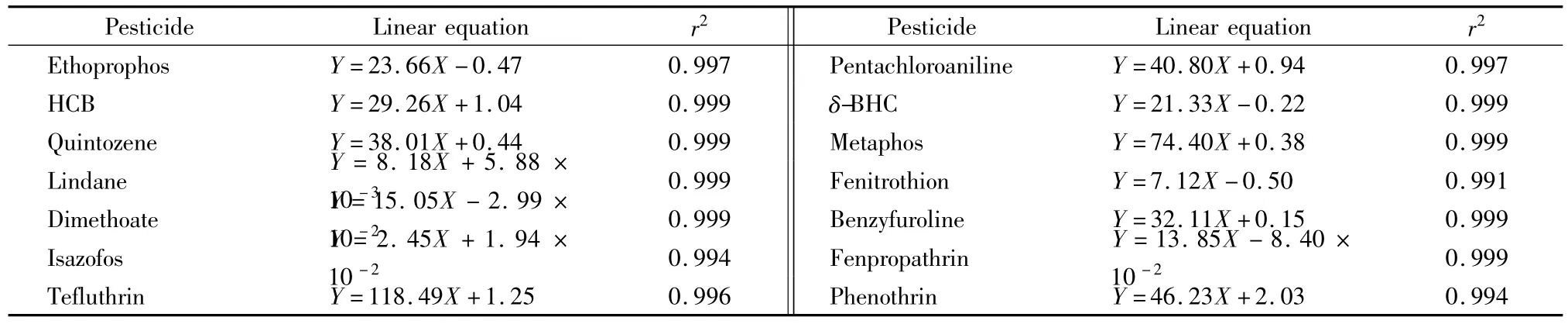
表3 14种农药的线性方程和相关系数Table 3 Linear equations and the correlation coefficients(r2)of the 14 pesticides
2.6 方法的准确度和精密度
以鲫鱼作为实际样品,分别在其中添加0.01、0.05、0.1 mg/kg 3个水平进行加标回收试验。采用内标法进行定量,内标物为环氧七氯。每个水平分别做6个平行样品,加标回收率和相对标准偏差(RSD)见表4。结果表明:3个加标水平的回收率为83.0%~112.9%,RSD为3.2%~12.0%,表明方法的准确度和精确度较高,满足检测需求。
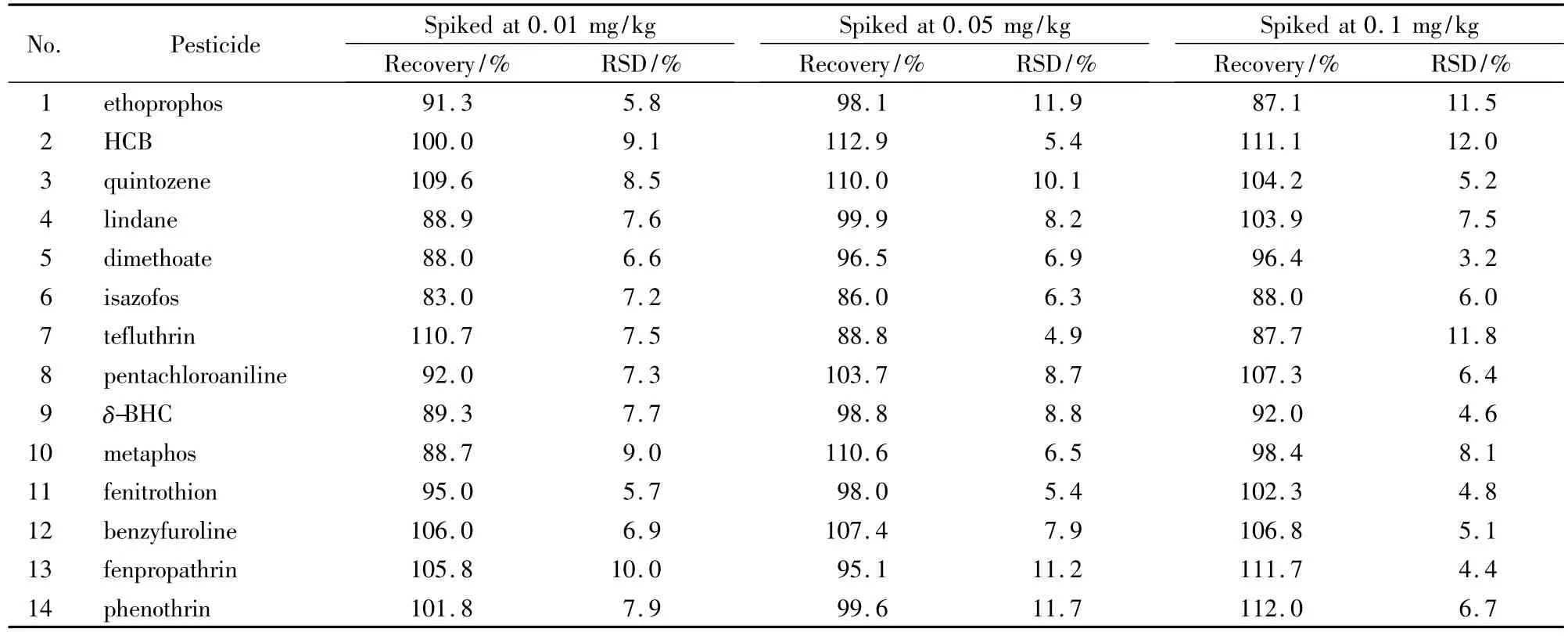
表4 鲫鱼样品中14种农药在3个加标水平下的回收率和相对标准偏差Table 4 Recoveries and RSDs of the 14 pesticides spiked in crucian carp at three levels
3 结论
建立了在线GPC-MDGC/MS检测动物源性食品中多种农药残留的方法。采用在线GPC净化方式可以有效地去除色素、脂肪等相对分子质量较大的物质的干扰。同时在线GPC缩短了分析时间,降低了有机溶剂的消耗。二维色谱分离模式可以使一维色谱无法分离的组分通过切割的方式进入二维色谱进一步分离,提高了分离效率,为多残留分析提供了可能。方法实现了在线GPC和二维色谱的有效结合,准确度好、精密度高,具有很好的推广性。
[1]Rimkus G G,Rummler M,Nausch I.J Chromatogr A,1996,737(1):9
[2]Fontcuberta M,Arqués J,Villalbí J R,et al.Sci Total Environ,2008,389(1):52
[3]Mukherjee I,Gopal M.J Chromatogr A,1996,754(1/2):33
[4]Saito K,Sjodin A,Sandau C D,et al.Chemosphere,2004,57(5):373
[5]Zhu W W,Allaway J R.J Chromatogr A,2004,1055(1):191
[6]Venkatesan M I,Northrup T,Phillips C R.J Chromatogr A,2002,942(1):223
[7]Yu S,Xu X M.Rapid Commun Mass Spectrom,2012,26(8):963
[8]Wu G,Bao X X,Wang H X,et al.Chinese Journal of Chromatography(吴刚,鲍晓霞,王华雄,等.色谱),2008,26(5):577
[9]Liu L B,Hashi Y,Qin Y P,et al.J Chromatogr B,2007,845(1):61
[10]Xu X M,Yu S,Li R,et al.Food Chem,2012,135(1):161
[11]Ouyang Y F,Tang H B,Wu Y,et al.Chinese Journal of Chromatography(欧阳运富,唐宏兵,吴英,等.色谱),2012,30(7):654
[12]Yang L X,Li H L,Zeng F G,et al.J Agric Food Chem,2012,60(8):1906
[13]Wu G,Bao X X,Zhao S H,et al.Food Chem,2011,126(2):646
[14]Cunha S,Fernandes J O.Talanta,2010,83(1):117
[15]Castillo M L R,Rodriguez-Valenciano M,Moreno F,et al.Talanta,2012,89(1):77
