单个砷原子在砷化镓富砷表面的迁移行为
2014-10-16杜诗文
李 坤,杨 雯,杜诗文
(1.太原科技大学应用科学学院,太原 030024;2.太原科技大学材料科学与工程学院,太原 030024)
砷化镓作为一种典型的Ⅲ-Ⅴ族半导体化合物,具有闪锌矿结构。GaAs体材料中每个Ga原子与周围的四个As原子成键,同样的,每个As原子也与周围的四个Ga原子成键。由于在GaAs体材料的表面,原子的配位数发生变化,所以处于GaAs表面的原子(As原子或是Ga原子)会产生部分不饱和悬挂键。正是由于这些悬挂键的出现,使得GaAs的表面具有了丰富多彩的再构形式[1-2]。
在GaAs材料取向众多的表面当中,GaAs(001)表面有着特殊的重要性。一方面是因为具有(001)表面的GaAs材料是一种广泛应用于光电器件的基础材料[3]。另一方面,现在获取GaAs材料最广泛使用的方法是逐层生长合成法(layer-by-layer synthesis),该方法得到的GaAs材料的表面一般是(001)表面[4]。即使只在(001)取向的表面上,GaAs的再构形式也是非常丰富的。在不同的实验条件下会呈现例如富砷(As-rich)的c(4×4),2×4,c(2×8),1×6,4×6,富镓(Ga-rich)的4×2等不同的再构形式[5-6]。而在这些再构形式中,研究最多的是β2(2×4)As-rich表面再构,这是由于该表面再构是 GaAs(001)表面诸多再构形式中最稳定的[7]。
在关于GaAs表面的研究工作中,原子在GaAs表面的迁移行为是一个重要的研究方向。这是由于在用分子束外延(MBE)生长过程中,将不可避免的遇到原子在表面迁移的情况。而对原子迁移行为的研究能够更深入的理解材料表面的基本性质。近年来的研究表明,在GaAs(001)β2(2×4)的表面上往往会吸附一些As原子(adatom As)或Ga原子(adatom Ga),这些吸附原子会沿着材料表面的某一路径进行迁移,并最终会聚合成具有稳定结构的原子团簇[8]。这一有趣的现象引起了人们广泛的关注。以往对GaAs表面原子迁移问题的研究大多是实验研究,而对于这方面的理论研究并不多。
本文利用经验势模型结合分子动力学的方法,研究了单个砷原子在GaAs(001)β2(2×4)As-rich表面的迁移行为。
1 研究方法
使用经验势模型模拟Ga-As系统,需要处理Ga-Ga、As-As以及 Ga-As等三种相互作用,Ga-As之间的相互作用主要是通过共价键结合,而As-As、Ga-Ga之间的相互作用则是通过混合的金属与共价键结合而成的。因此,对于经验势模型来讲,想要准确的模拟Ga-As体系,至少应能够处理金属键和共价键这两种成键形式。本文所采用的经验势模型,是由Albe等人发展的键序势模型[9],该模型采用了一种短程的键序势来模拟金属键和共价键这两种相互作用。这种经验势模型成功的模拟了包括GaAs在内的绝大多数III-V族化合物,其结果得到了人们的广泛认可。
根据势模型,体系的总能E可表示为:
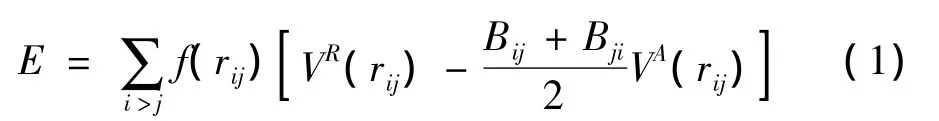
上式中VR和VA分别为排斥项和吸引项:
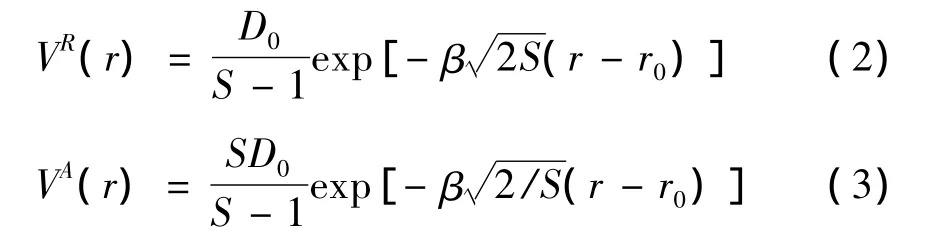
其中,D0是原子对的能量,r0是原子对的平衡键长,S是一个可调节参数。参数β是与原子对基态振动频率相关的参数:
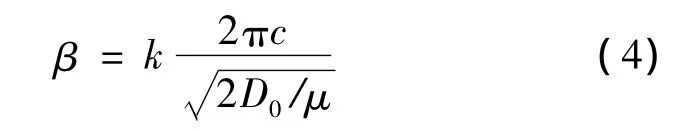
其中,k是原子对基态振动的波数,μ是折合质量,c是光速。
式(1)中f(r)是原子间相互作用的截断函数:
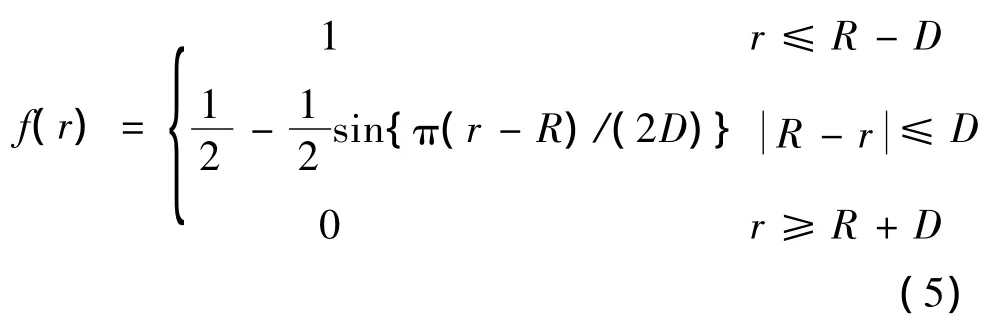
其中,D和R是可调节参数。若两原子间的距离r≥R+D,则两原子间相互作用为零。
式(1)中Bij是含有角度项的键序参量,用于模拟原子间的相互作用随键角和键长的变化,采用了Brenner等人给出的形式[10]:
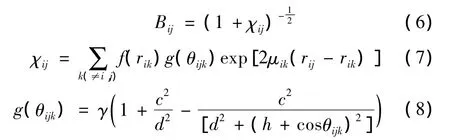
其中,μ,c,d和h都是可调参数。
结合以上的键序势模型,采用分子动力学方法计算了单个砷原子在砷化镓(001)β2(2×4)富砷表面迁移的势能面,在此基础上分析了砷原子在该表面上的迁移行为。
2 As原子在GaAs表面的迁移
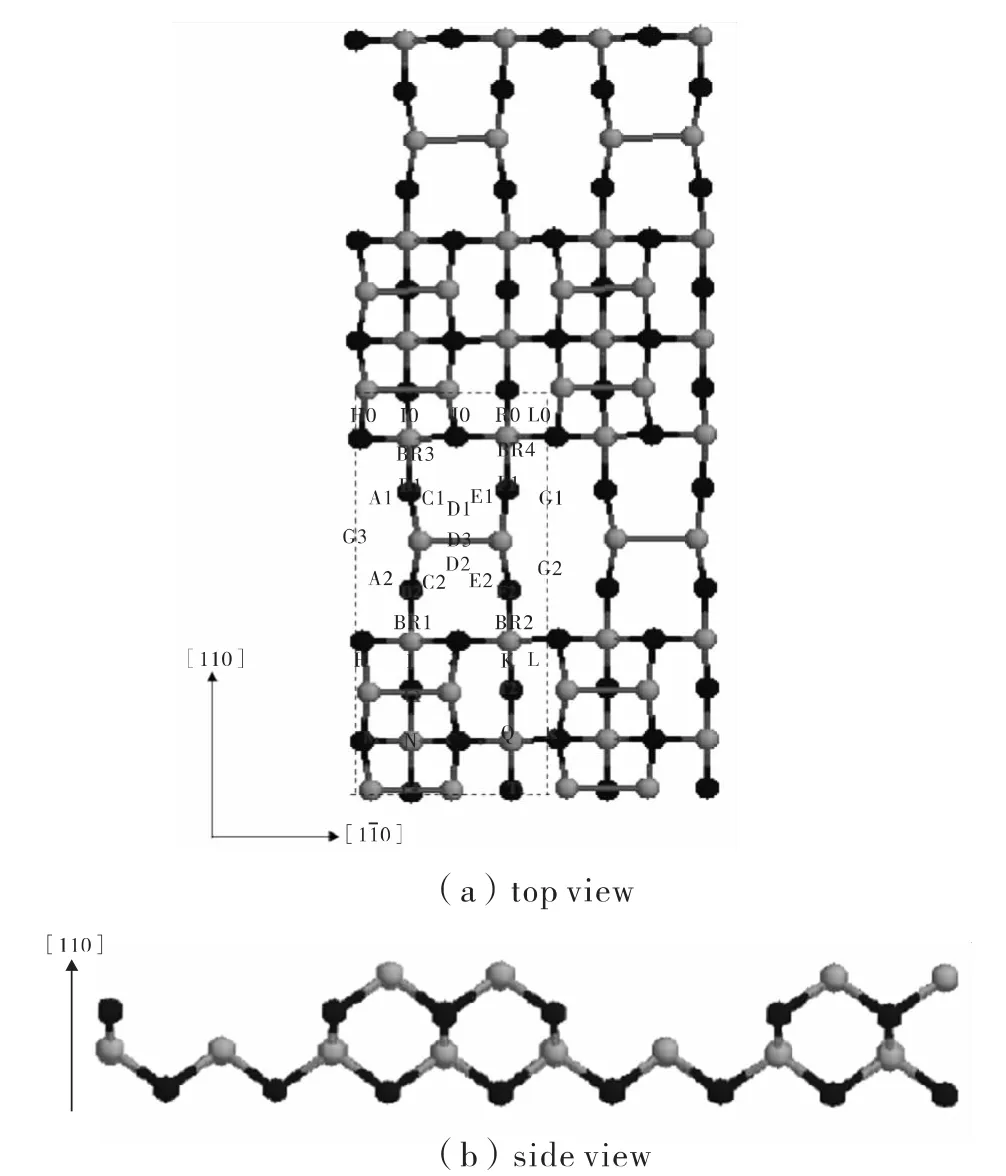
图1 (a)GaAs(001)β2(2×4)As-rich表面俯视图,浅色球为As原子,深色球为Ga原子,虚线部分是一个原胞。(b)GaAs(001)β2(2×4)As-rich表面侧视图Fig.1 (a)Top view of GaAs(001)β2(2 ×4)As-rich surface.The light spheres were As atoms and the dark spheres were Ga atoms.(b)Side view of GaAs(001)β2(2×4)As-rich surface
GaAs(001)β2(2×4)As-rich表面的结构如图1所示,其几何构型是最上层有两个砷二聚体(Asdimer),在第三层有一个 A s-dimer[11-12]。在计算过程中,分别在该表面的(110)方向和方向取两个周期,从而组成一个超原胞(图1(a)).同时在(001)方向上取18个原子层,且在原子层上方取50厚的真空层,由此确保吸附原子之间、上下表面之间都不具有相互作用。为方便起见,我们把(001)方向设为z方向,把(110)方向和方向分别设为(x,y)方向。为了得到单个As原子在该表面的势能面(Potential Energy Surface,PES),沿(110)方向和将表面原胞(图2(a)中虚线部分)均匀的划出相隔为 0.2的格点。然后将As原子放在这些格点上方,距离表面3.0的位置。固定吸附原子的x方向与y方向,只让其在z方向上有一个自由度。最后,结合第二部分所介绍的键序势模型,利用分子动力学方法对整个体系的总能进行模拟计算。因此可以得到吸附原子吸附在(x,y)点时体系的总能E(x,y).以能量最低的E(x,y)作为参考能量(即零点),进而可以得到相对吸附能的PES(图2).
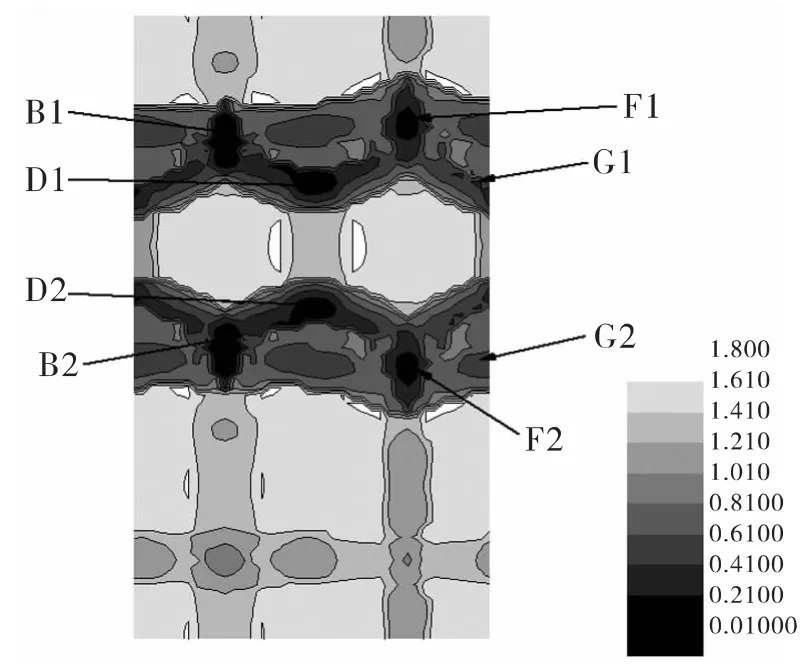
图2 单个As原子在GaAs(001)β2(2×4)As-rich表面的势能面(PES),每条等高线之间的差距是0.2 eV,深色的区域表示能量较低的区域,字母标出的位置与图1中位置相对应Fig.2 Potential Energy Surface(PES)of an extra As atom on the GaAs(001)β2(2 ×4)As-rich surface,The unit of the isohypse line was 0.2 eV,where the dark area presented adsorption positions with lower potential energy.The positions were identical to those in Fig.1.
在PES图中,存在一些特殊区域,在这些区域里,原子的相对吸附能要比周围区域的低。我们把这些区域称为迁移通道或迁移路径,即原子容易在这些区域吸附或沿着这些区域迁移。因此原子更容易在迁移路径上聚合,从而形成团簇。在图2中,D1点是As原子在整个表面上相对吸附能最低的点。在该点,吸附As原子与两个As原子和一个Ga原子成键,形成三配位的情况。此外,发现As原子在GaAs(001)β2(2×4)As-rich表面迁移的过程中,存在两条迁移路径,分别是路径(A1-B1-C1-D1-E1-F1-G1)和路径 (A2-B2-C2-D2-E2-F2-G2),如图2所示。而这两条路径对称的分布在表面的台阶附近,情况完全相同,实际上是一条路径。几乎所有相对吸附能较低的点都集中在该路径上。而且在整个路径上,相对吸附能都很低(低于0.6 eV),如图3所示。这意味着即使是在温度很低的情况下,As原子也有机会沿着该迁移路径进行迁移。而且在室温下,As原子有可能在该迁移路径上形成团簇。

图3 As原子在GaAs(001)β2(2×4)As-rich表面沿路径(A1-B1-C1-D1-E1-F1-G1)迁移时的相对吸附能。字母表示吸附位,位置与图1相对应Fig.3 The relative adsorption energy of an extra As atom diffusing along the path(A1-B1-C1-D1-E1-F1-G1)on the GaAs(001)β2(2 ×4)As-rich surface.The positions were identical to those in Fig.1.
在不同吸附位有不同的相对吸附能,主要是由于在不同的吸附位上吸附As原子与周围原子的成键情况是不同的。对比在迁移路径上吸附原子与周围原子的成键情况,发现当As原子在迁移路径上迁移时,其成键配位数是二、三交替的。在能量较低的点B1,D1,F1,G1,吸附原子与周围原子是三配位成键。其中在B1和F1点吸附时,As原子与一个As原子和两个Ga原子成键。在D1和G1点吸附时,As原子与两个As原子和一个Ga原子成键。在吸附能较高的点A1,C1,E1,As吸附原子与一个As原子和一个Ga原子形成两配位的情况。

表1 As原子在GaAs(001)β2(2×4)As-rich表面沿迁移路径迁移时的平均键长和平均键角Tab.1 Mean bond length and bond angle of an extra As atom diffusing along the path on the GaAs(001)β2(2×4)As-rich surface
接下来,进一步分析了在迁移路径上的吸附As原子与周围原子的构型。表1中统计了吸附As原子在迁移路径上迁移时的平均键长和平均键角。结果表明,As-Ga键的平均键长 2.48,接近于GaAs晶体中的 Ga-As键的键长(2.447).另外,当As原子在迁移路径上迁移时的平均键角111°也接近于GaAs晶体中的值109°28'.因而 As原子在迁移路径上迁移时,所受张力非常小。这进一步解释了吸附As原子更倾向于在迁移路径上迁移的原因。
3 结论
通过以上的模拟计算和分析讨论,我们发现As原子在GaAs(001)β2(2×4)As-rich表面上进行迁移时,存在一条沿着表面台阶处、平行于As二聚体的迁移路径。通过分析As原子在迁移路径的各吸附位上与周围原子的成键形式和构型,发现当吸附As原子与周围原子形成与GaAs晶体结构近似的四面体结构时相对吸附能较低,即形成的构型更稳定。进而解释了As原子倾向于在迁移路径上迁移的原因。此外,As原子在沿着该迁移路径进行迁移的过程中,所需克服势垒很小(小于0.6 eV),因此在室温下As原子有可能沿着该路径进行迁移,并聚合为团簇。该迁移路径的发现,可以为相应的实验和应用提供理论指导和参数依据。
[1]OHTAKE A.Surface reconstructions on GaAs(001)[J].Surf Sci R,2008,63:295-327.
[2]XUE Q K,HASHIZUME T,SAKURAI T.Scanning tunneling microscopy ofⅢ-Ⅴ compound semiconductor(001)surfaces[J].Prog Surf Sci,1997,56(1-2),1-131.
[3]TERESHCHENKO O E,CHIKICHEV S I,TEREKHOV A S.Composition and structure of HCl-isopropanol treated and vacuum annealed GaAs(100)surfaces[J].J Vac Sci Technol A,1999,17(5):2655-2662.
[4]THOMAS J C,VEN A V,MILLUNCHICK J M.Considerations for surface reconstruction stability prediction on GaAs(001)[J].Phys Rev B,2013,87(7):075320.
[5]HASHIZUME T,XUE Q K,ZHOU J,et al.Structures of As-Rich GaAs(001)-(2 × 4)Reconstructions[J].Phys Rev Lett,1994,73(16):2208-2211.
[6]HASHIZUME T,XUE Q K,ICHIMIYA A,et al.Determination of the surface structures of the GaAs(001)-(2 × 4)As-rich phase[J].Phys Rev B,1995,51(7):4200-4212.
[7]THOMAS J C,MODINE N A,MILLUNCHICK J M.Systematic approach for determination of equilibrium atomic surface structure[J].Phys Rev B,2010,82(16):165434.
[8]TSUKAMOTO S,KOGUCHI N.Magic numbers in Ga clusters on GaAs(001)surface[J].J Cryst Growth,2000,209(2-3):258-262.
[9]ALBE K,NORDLUND K,AVERBACK R S.Modeling of compound semiconductors:Analytical bond-order potential for Ga,As,and GaAs[J].Phys Rev B,2002,66(3):035205.
[10]BRENNER D.Relationship between the Embedded-Atom Method and Tersoff Potentials[J].Phys Rev Lett,1989,63(9):1022.
[11]NORTHRUP J E,FROYEN S.Structure of GaAs(001)surfaces:The role of electrostatic interactions[J].Phys Rev B,1994,50(3),2015-2018.
[12]LABELLA V P,YANG H,BULLOCK D W,et al.Atomic Structure of the GaAs(001)-(2 ×4)Surface Resolved Using Scanning Tunneling Microscopy and First-Principles Theory[J].Phys Rev Lett,1999,83(15):2989-2992.
