Ni-Mo合金电沉积规律研究*
2014-09-18陈范才刘雪江唐兴宇王淼宇
陈范才, 刘雪江,徐 超,唐兴宇,王淼宇
(湖南大学 化学化工学院,湖南 长沙 410082)
氢能是一种清洁、可再生的二次能源,将会成为未来主要的能源之一.而决定氢能能否被广泛使用的关键在于制氢所使用的阴极材料的性能,故大量的工作致力于获得一种高催化活性、低析氢过电位的析氢电极材料.Ni-Mo合金作为一种耐腐蚀性强、析氢过电位极低的优良阴极材料而受到极大的关注.Ni-Mo合金的制备方法多种多样,其中,电沉积方法制备的Ni-Mo合金性能优良、工艺成熟、成本低廉,故人们对Ni-Mo合金的电沉积制备工艺[1—3]进行了大量的研究,目前以含氨水的碱性柠檬酸盐溶液中电沉积制备的Ni-Mo合金的工艺最为成熟,而关于Ni-Mo合金的沉积机理研究[4—7]也取得了一定的进展,目前为人们普遍接受的沉积机理为诱导共沉积机理.但Ni-Mo合金共沉积过程中的规律并未得到过系统的研究,本文通过恒电位沉积得到的极化曲线和电流效率、CV测试来探究Ni-Mo合金的共沉积规律.
1 实验部分
1.1 溶液与实验仪器
实验溶液所用水均为二次去离子水,试剂均为分析纯,各溶液组成如表1所示.测试系统为CHI760B电化学工作站,参比电极为Hg/HgO电极,辅助电极为大面积铂片,工作电极为99.99%Pt电极,面积为2.0 cm2×2,使用前先用7#金相砂纸打磨,然后依次用丙酮、二次去离子水清洗,备用.

表1 实验溶液组成
1.2 恒电位沉积
以0.05 V为间隔在-0.7~-1.3 V(vsHg/HgO)取13个电位值恒电位沉积80 min,温度为25 ℃,采用紫外分光光度法[8]测定Pt电极上沉积的Ni,Mo含量,再通过计算得到平均沉积电流I(Q=zFm/M,I=Q/t,Q为沉积电量,z为金属还原消耗电子数,m为金属的质量,M金属的摩尔质量,t为时间)和电流效率η(η=Im/I,Im为金属还原电流,I为总电流),取各电位下的I和η值绘图得到极化曲线和电流效率图.
1.3 循环伏安测试
在C溶液基础上改变其中Ni2+的含量;在D溶液基础上改变Ni2+的含量而保持MoO42-的含量不变,改变MoO42-的含量而保持Ni2+的含量不变.进行循环伏安测试,调整扫描速度为0.01 V/s,扫描电位范围为0~-1.5 V.
2 结果与讨论
2.1 4种溶液阴极极化曲线
图1为Pt电极在A,B,C和D 4种溶液中的总极化曲线.由于溶液A中阴极不会发生任何金属的沉积,只有析氢,阴极电流都用于氢的还原,曲线a应为碱性柠檬酸盐溶液中Pt电极的析氢极化曲线.由曲线b可知,Pt电极在溶液B中电流随电位的变化规律与A溶液基本一致,且电极上未检测到金属Mo,溶液B中Mo的电沉积电流为零,可见Mo不可能单独从碱性含氨的柠檬酸盐水溶液中还原为金属.曲线c位于曲线a下方,这是由于溶液C中发生了过电位较高的Ni的沉积,而Ni覆盖在Pt电极表面又降低了析氢催化活性,从而使得阴极极化增大、电流减小.在溶液D中,Ni-Mo共沉积并覆盖在Pt电极表面,MoO42-降低了Ni的沉积过电位同时Ni可诱导MoO42-的还原而发生Ni-Mo共沉积,Ni-Mo合金具有高效的析氢催化活性,使得阴极极化降低、电流大增,故曲线d位于曲线a上方.

E/V
2.2 Ni单独沉积时的极化行为
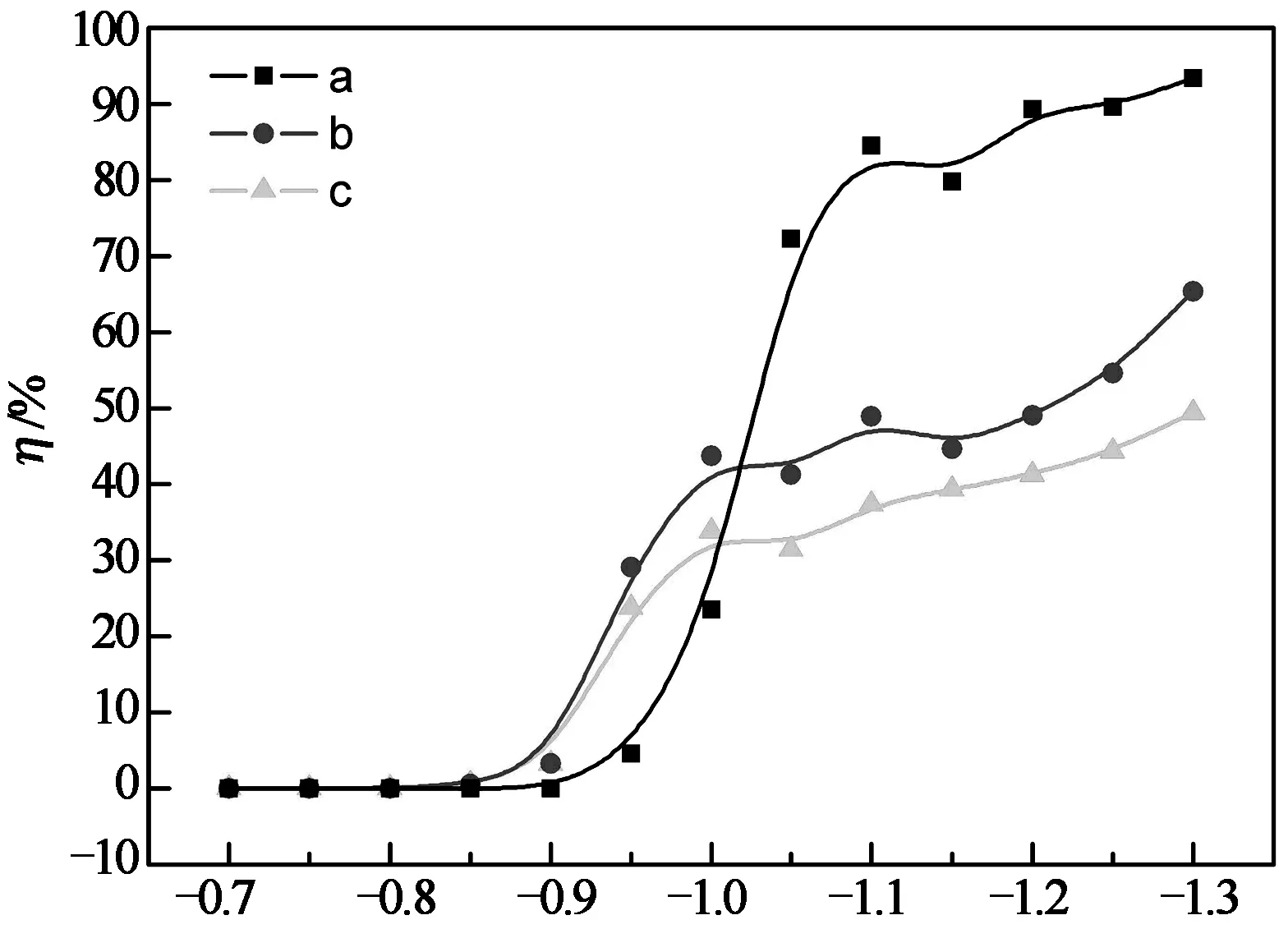
图2为Pt电极在溶液C中的总极化曲线和分支极化曲线以及电流效率随电位的变化关系.从图2中Ni的分支极化曲线可知,Ni在-0.9 V左右开始沉积,在-0.9 V>E>-1.0 V区域,极化电流较小的Ni的沉积非常缓慢,此时阴极电流主要用于氢的析出.在E<-1.0 V时,Ni的沉积电流迅速增加,此时Ni优先沉积,Ni的沉积速度远大于氢的析出速度.由图中Ni的电流效率曲线可以发现Ni的沉积效率在-1.0 V>E>-1.1 V时迅速增大,当电位达到-1.1 V时,电流效率较高且逐渐趋于稳定.可见溶液C中Ni在较低电位下的沉积效率较高.
图3为改变溶液C中Ni2+浓度得到的循环伏安曲线,从图中可知Ni的沉积有两个还原峰,Ni2+的还原是分两步进行[7]的,即:
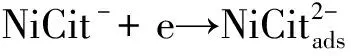
(1)

(2)
从该图中还可以发现随Ni2+含量的增加,第一个还原峰电位正移,随着溶液中Ni2+浓度增加,反应(1)的电位E=E+(RT/zF)lna(NiCit-)中的lna(NiCit-)也增大,从而使得反应(1)的电位正移.

E/V

E/V
2.3 Ni-Mo共沉积时的极化行为


E/V
图5为溶液D中MoO42-含量变化对Ni-Mo沉积的影响,由图5可知在溶液D中并没有出现与溶液C类似的双峰,只有一个还原峰,同时从图中还可知随MoO42-含量的增加合金的还原峰电位发生负移,可见溶液D中MoO42-的含量变化影响了合金沉积电位.图6为溶液D中Ni2+含量的变化对峰电位的影响,由图6可见,溶液D的还原峰电位也随Ni2+含量增加而正移,这与溶液C的变化规律一致.
图7为Pt电极在溶液C和D中的电流效率随电位的变化,对比曲线a和c可以发现在-0.85 V>E>-1.03 V时,溶液D中Ni沉积电流效率大于溶液C,而在E<-1.03 V时D溶液中Ni的沉积电流效率小于C溶液.同样,对比曲线a,b可以发现类似的规律.而由图8可知溶液D中Ni沉积电流在E>-1.1 V时大于溶液C,在E<-1.1 V时小于溶液C.综上可见,MoO42-的加入降低了Ni的沉积过电位,但随着Mo的开始沉积,合金共沉积的总电流效率增长缓慢且随电位负移而低于溶液C中Ni的沉积效率,这可能与Mo6+的沉积机理有关:
MoO42-+2H2O+2e→MoO2+4OH-
(3)

(4)

(5)

E/V
E/V

E/V
3 结 论
1)在碱性柠檬酸盐溶液中, MoO42-的加入可以使Ni-Mo合金中Ni更容易沉积出来,比只含Ni2+溶液中Ni沉积电位正移了0.05 V,但降低了Ni在高过电位下的沉积电流效率,同时也使得Ni-Mo合金的电流效率较低,Ni-Mo合金电流效率总和在低电位下比溶液C中Ni沉积效率低约20%.
2)Ni沉积的第一个峰电位及Ni-Mo合金还原峰电位均随溶液中Ni2+含量的增加而逐渐正移,Ni-Mo合金沉积峰电位随MoO42-含量增加而发生负移.
[1]LUCIANA S S, SERGIO H D, CLAUDIAE B E,etal. Characterization of electrochemically deposition Ni-Mo alloy coatings[J].Electrochemistry Communications,2004,6:543-548.
[2]曾跃,姚素薇,郭鹤桐.非晶态Ni-Mo合金电沉积[J].电镀与精饰,1994,16(3):9-12.
ZENG Yue, YAO Su-wei, GUO He-tong. Electrodeposition of amorphous Ni-Mo Alloy[J].Plating & Finishing,1994,16(3): 9-12. (In Chinese)
[3]LI Ning,CHEN Wei-zeng,JING Bao-de,etal. Preparations and properties of amorphous/nanocrystal Ni-Mo alloys coating obtained by electrodeposition[J]. Advanced Materials Research, 2011,148/149:674-678.
[4]CHASSAING E, QUANG K V. Mechanism of nickel-molybdenum alloy electrodeposition in citrate electrolytes [J].Journal of Applied Electrochemistry,1989,19:839-843.
[5]UEKAWA E, MURASE K, MATSUBARA E,etal. Determination of chemical species and their composition in Ni-Mo alloy plating baths by factor analysis of visible adsorption spectra[J].Journal of the Electrochemical Society,1998,145(2):523-528.
[6]LI Ning, GAO Cheng-hui, CHENG Guang-ming. Preparations effects on amorphous/nanocrystal Ni-Mo alloys structure and properties[J].Advanced Materials Research,2011,217/218:21-26.
[7]PODLAHA E J,LANDOLT D. Induce codeposition, Ⅲ. Molybdenum alloys with nickel, cobalt and iron[J].Journal of the Electrochemical Society,1997 ,144(5):1672-1680.
[8]黄亮.泡沫镍钼合金电极材料制备工艺的设计、研究与中试[D].长沙:湖南大学,2012:26-36.
HUANG Liang. The design, research and pilot study on the preparation process of foamed nickel molybdenum alloy electrode material[D].Changsha: Hunan University,2012:26-36.(In Chinese)
