鸡胚胎干细胞多能性相关基因cNanog和cPouV干扰载体的构建及筛选
2014-09-14彭特王丙云李东升陈志胜陈胜锋计慧琴陈金顶
彭特 王丙云 李东升 陈志胜 陈胜锋 计慧琴 陈金顶
在哺乳动物和人中,Oct4、Nanog和Sox2被认为是调控干细胞多能性及自我更新的3个重要的转录因子[1-3],但它们在家禽干细胞中的作用存在争议。2001年Soodeen等[4]研究表明,鸡体内不存在Oct4,Lavial等[5]的研究发现鸡胚胎干细胞表达Oct4同源基因鸡PouV(cPouV),并且对其多能性的调控有重要作用。本实验室的前期研究显示,在鸡早期胚胎发育过程中,多能性相关基因cNanog、cPouV和Sox2显著表达,但随着胚胎发育3个基因的表达量呈明显的下降趋势,在鸡胚胎干细胞分化的过程中,cNanog、cPouV和Sox2的表达水平也显著下降,导致细胞多能性的丧失,提示这3个基因在维持鸡胚胎干细胞多能性中发挥重要作用[6-8]。
RNA干扰(RNA interference,RNAi)是双链RNA(dsRNA)介导的遗传干扰现象,它能够特异有效地降解mRNA,从而引起转录后水平的基因沉默,导致相应功能基因表型缺失[9]。1998年首次在秀丽线虫中发现RNA干扰现象,不久后在真菌、植物、果蝇、锥虫、涡虫、水螅、斑马鱼等真核生物中都发现了RNA干扰现象[10,11],RNA干扰技术具有高效、快速和成本低廉的优势,现已经成为研究基因功能、新基因筛选、基因治疗和寻找药物靶点的重要工具[12,13]。
为进一步阐明cNanog和cPouV基因在鸡胚胎干细胞多能性维持和自我更新中的作用,本试验分别针对鸡多能性相关基因cNanog和cPouV设计3对siRNA干扰片段,合成shRNA插入到干扰载体中,构建siRNA表达载体,并对其进行筛选,以期获得良好的干扰载体和转染技术,为获得cNanog和cPouV基因稳定干扰的CES细胞株,阐明cNanog和cPouV基因在鸡胚胎干细胞多能性调控中的作用奠定基础。
1 材料与方法
1.1 材料
新鲜受精鸡蛋(麻黄鸡)购于广东省佛山市南海种禽有限公司;pCMV-N-Flag、pSuper-Retro-Puro和pEGFP-N1质粒由广州医科大学蔡铭升博士惠赠;实验所用的限制性内切酶、DNA Marker、逆转录试剂盒和RNAiso Plus均为大连TaKaRa公司产品;T4DNA连接酶为NEB公司产品;KOD酶购自东洋纺公司;质粒小量提取试剂盒和DNA凝胶回收试剂盒为上海生工公司产品;DYKDDDDK-Tag Mouse mAb和Actin Mouse mAb为Abmart公司产品;山羊抗小鼠IgG(H+L)和超敏ECL化学发光试剂盒购自碧云天研究所;PVDF膜购自Millipore公司;寡核苷酸和引物由Invitrogen公司合成;测序由上海生工公司完成。
1.2 方法
1.2.1 cNanog和cPouV基因 siRNA 表达载体的构建 根据siRNA的设计原则,通过Ambion公司在线siRNA靶位点设计软件(http://www.ambion.com/techlib/misc/siRNA_tools.html)针对 cNanog(NM_001146142)和 cPouV(NM_001110178)基因分别设计3条寡聚核苷酸序列(表1)。合成的寡聚核苷酸配制成100μmol/L的浓度,与互补的寡聚核苷酸片段混合后用退火缓冲液(100mol/L NaCl、pH7.450mmol/L HEPES)稀释成终浓度3pmol/L。90℃4min,70℃ 10min孵育,缓慢冷却至室温。退火后形成的shRNA连接到经Bgl II和Hind III酶切的pSuper-Retro-Puro质粒中,连接产物转化DH5α感受态细胞、挑取克隆摇菌后送测序公司测序。提取测序正确的干扰载体,保存用于后续试验,分别命名pS1-cNanog、pS2-cNanog、pS3-cNanog和 pS1-cPouV、pS2-cPouV、pS3-cPouV。

表 1 合成的用于构建干扰载体的寡核苷酸序列
1.2.2 cNanog和cPouV全基因表达载体的构建 用Clone Manager软件设计扩增cNanog和cPouV基因的特异性引物,分别在扩增cNanog基因的引物引入BamH I和EcoR I,cPouV基因引入EcoR I和Sa lI酶切位点(表2)。采用本实验室改进的方法取新鲜种鸡蛋的胚盘[14],按照说明书采用Trizol法提取总RNA,并用AMV逆转录酶和Oligo(dT)18引物合成cDNA,以合成的cDNA为模板按照下列参数进行PCR 扩增 :94℃ 5min;94℃ 30s,退火 30s,72℃50s,循环35次;最后72℃ 10min。PCR 产物回收纯化后用双酶切,回收目的片段与经同样酶切及回收的pCMV-N-Flag载体连接,转化后挑选阳性菌落进行PCR与酶切鉴定以及测序验证,将构建正确的重组质粒分别命名为pCMV-Flag-cNanog和pCMVFlag-cPouV。

表2 cNanog和cPouV基因的PCR引物序列
1.2.3 pSuper-shRNA、pCMV-Flag重组表达载体和pGFP质粒共转染293T细胞 采用磷酸钙法转染293T细胞,转染前1d选择对数生长期的293T细胞接种于6孔板,待细胞生长至80%汇合时取4μg pSuper-shRNA、4μg pCMV-Flag 重 组 质 粒 和 0.5μL pGFP-N1质粒加入到100μL CaCl2溶液中,混匀,将混合液缓慢滴加入100μL 2×HBS中,剧烈混合后加入6孔板中,继续培养细胞。
1.2.4 Western blot法检测重组载体的干扰效果 3个siRNA表达载体和全基因表达载体以及pGFP质粒共转染293T细胞24h后,观察荧光以确定转染效率,然后提取细胞蛋白进行Western印迹,以未转染siRNA表达载体组为阴性对照,β-actin作为内源性对照。
293T细胞用预冷的PBS洗涤两次,6孔板按100μL/孔加细胞裂解液,在冰上震荡20min后重悬,12000g 4℃离心5min。经SDS-PAGE分离后,电转移至PVDF膜上。膜封闭后分别用抗Flag(1∶3000),抗 β-actin(1∶3000)抗体 4℃孵育过夜,与辣根过氧化物酶标记的羊抗鼠二抗(1∶1000)室温反应1h,用化学发光法检测目标基因在干扰前后的表达变化。
2 结果
2.1 siRNA 表达载体的构建
用pSuper-Retro-Puro质粒为基础质粒进行siRNA表达载体的构建,载体含有氨苄抗性基因,表达载体连接产物转化大肠杆菌DH5α感受态细胞,涂布于LB氨苄青霉素抗性平板培养过夜,挑取单克隆培养并提取质粒,重组质粒测序验证与预期相符,部分测序结果见图1。

图1 重组质粒pS3-cPouV的测序比对图
2.2 cNanog和cPouV全基因表达载体构建与鉴定
经PCR扩增cNanog和cPouVcDNA编码区全长,1%琼脂糖凝胶电泳检测,结果(图2)显示扩增出长约946bp的cNanog和905bp的cPouV目的片段;将重组质粒pCMV-Flag-cNanog和pCMV-Flag-cPouV分别用BamH I、EcoR I和EcoR I、Sal I进行双酶切后,电泳图谱显示分别切出与插入的预期目的大小相符的DNA 条带(图3)。进一步的测序结果表明,插入片段的核苷酸序列与GenBank公布的cNanog和cPouV参考序列完全相同,表明重组质粒构建成功。
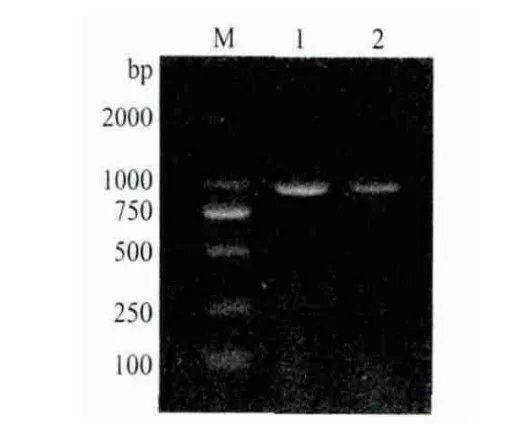
图2 cNanog和cPouV目的片段PCR产物电泳图谱
2.3 质粒转染293T细胞效率的估测
载体转染24h后,荧光显微镜下检测 pGFP转染效果,可见约70%-80%的293T细胞发出绿色荧光(图4),由于共转染时加入的pGFP质粒的浓度及体积一样,从侧面可反映出重组干扰载体的转染效率情况。表明磷酸钙介导的重组干扰载体转染293T细胞效率较高且不同质粒间无明显差异。上述试验独立重复5次以上,结果均一致。
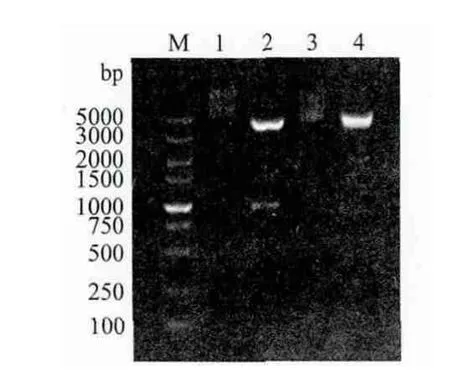
图3 pCMV-Flag重组质粒双酶切产物电泳图谱
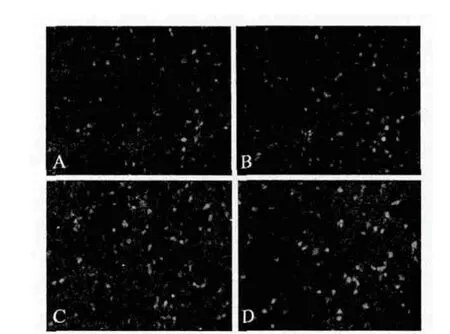
图4 GFP与不同质粒共转染293T细胞表达情况(24h,100×)
2.4 cNanog和cPouV 干扰片段效率检测
分别用cNanog和cPouV基因的3种pSupershRNA和pCMV-Flag重组表达载体共转染293T细胞后,Western blot检测结果(图5)发现,所构建的重组干扰载体均有一定的抑制靶基因表达的作用。其中针对cNanog基因的干扰载体中pS3-cNanog的抑制效果最明显,pS2-cNanog次之;pSuper-cPouV-shRNA中pS3-cPouV重组载体的效果最好,pS1-cPouV和pS3-cPouV也有一定的抑制作用。上述试验独立重复3次,结果均一致。
3 讨论
鸡胚胎干细胞在胚胎发育基础、转基因禽类的生产及家禽育种等方面具有巨大的应用前景,但难以实现体外长期培养以及可传代遗传修饰,究其原因是对其全能性维持机制缺乏深入的了解。研究表明cNanog和cPouV在调控胚胎干细胞的自我更新和维持多能性上起重要作用,其在哺乳动物中的研究取得了较多的成果,但在家禽中研究报道很少。利用RNAi技术研究鸡胚胎干细胞多能性相关基因的报道更是少之又少,在国内尚未有见。本试验成功构建了针对多能性相关基因cNanog和cPouV的重组干扰载体,以期进一步深入研究其在鸡胚胎干细胞中的生物学功能。
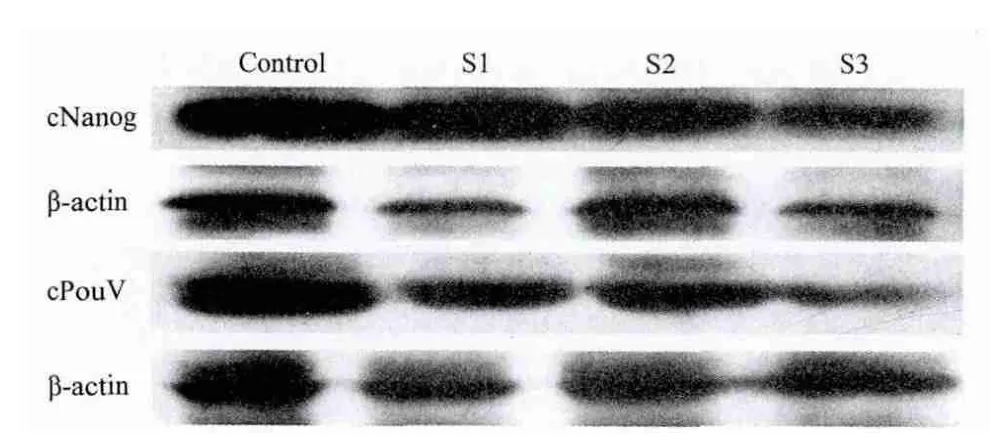
图5 转染pSuper-shRNA 24h后cNanog和cPouV蛋白表达水平
siRNA的获得方法有多种,包括化学合成、体外转录、长片段dsRNAs经RNaseIII类降解、siRNA表达载体或者病毒载体在细胞中表达、siRNA表达框在细胞中表达等,前3种方法的最大缺点是都需要直接操作RNA,操作不便,并且siRNA进入细胞后容易降解,RNAi效应维持时间短。为了克服这些不足,本研究采用了基于pSuper质粒表达siRNA的方式进行RNAi,不仅降低了制备siRNA的成本,简化了操作,而且延长了siRNA作用时间,便于筛选稳定表达细胞系用于研究基因的长期功能。pSuper质粒含有RNA聚合酶III识别H1启动子和一个5-6个连续的胸腺嘧啶(T)构成的转录终止位点以及选择标记[15]。本试验合成的寡核苷酸包含所选序列的正义和反义序列间用一段9nt的链(5'-TTCAAGAGA-3')连接;同时还包含5个连续的T作为RNA聚合酶III的终止信号,预测体内转录后会形成3'端两个U突出的茎部为19bp,环为9bp的短发夹结构,从而有效模拟RNA i过程中间体[16]。且pSuper质粒带有Puro标签,转染细胞后,可使用嘌呤霉素筛选稳定表达目的蛋白的细胞株。采用的真核表达载体pCMV-N-Flag的启动子CMV可以高效启动目的蛋白cNanog和cPouV在293T细胞中的表达,含有1个可以编码Flag标签的序列,可以表达出含有Flag标签的融合蛋白,便于使用抗Flag的抗体来识别目的蛋白,有利于目的蛋白检测。质粒转染细胞的方法有多种,如电穿孔法、磷酸钙法、脂质体法和病毒介导法等,本试验采取的磷酸钙转染法操作简便,易于得到稳定转染,但对pH值要求较高且重复性差,因此每种质粒都反复进行了多次转染以保持稳定,转染结果不仅进一步证实了表达载体pCMV-Flag和pSuper-Retro-Puro重组质粒可以转染293T细胞,而且具有较高的转染效率。
4 结论
本试验首先构建多能性相关基因cNanog和cPouV真核表达载体与pSuper-shRNA干扰载体,鉴定插入片段与预期相符后,共转染293T细胞以表达目的蛋白。再采用Western blotting对构建的重组干扰载体进行检测与筛选,在β-actin表达量保持相对一致时,加入重组干扰质粒组的靶基因蛋白表达量较对照组有所下降,表明构建的pSuper-shRNA能特异性抑制靶基因的表达,且不同位点的序列抑制效果不同。
[1]Rodda DJ, Chew JL, Lim LH, et al. Transcriptional regulation of nanog by OCT4 and SOX2[J]. J Biol Chem, 2005, 280(26):24731-24737.
[2]Mitsui K, Tokuzawa Y, Itoh H, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells[J]. Cell, 2003, 113(5):631-642.
[3]Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency[J]. Cell Res, 2007, 17(1):42-49.
[4]Soodeen-Karamath S, Gibbins AM. Apparent absence of oct 3/4 from the chicken genome[J]. Mol Reprod Dev, 2001, 58(2):137-148.
[5]Lavial F, Pain B. Chicken embryonic stem cells as a non-mammalian embryonic stem cell model[J]. Dev Growth Differ, 2010, 52(1):101-114.
[6]刘本杰, 陈志胜, 陈胜锋, 等. 全能性相关基因cNanog、cPouV和Sox2在鸡早期胚胎中的表达[J]. 中国组织工程研究, 2012(41):7733-7736.
[7]王丙云, 陈胜锋, 刘本杰, 等. 全能性相关基因在鸡胚胎干细胞中分化过程中的表达[C]. 南京:中国畜牧兽医学会动物生理生化学分会, 2012.
[8]陈志胜, 刘本杰, 王丙云, 等. 全能性相关基因在早期鸡胚发育过程中的表达[C]. 南京:中国畜牧兽医学会动物生理生化学分会, 2012.
[9]Fire A, Xu S, Montgomery M K, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans[J].Nature, 1998, 391(6669):806-811.
[10]Bosher JM, Labouesse M. RNA interference:genetic wand and genetic watchdog[J]. Nat Cell Biol, 2000, 2(2):E31-E36.
[11]Sharp PA. RNA interference--2001[J]. Genes Dev, 2001, 15(5):485-490.
[12]Tabara H, Grishok A, Mello CC. RNAi in C. elegans:soaking in the genome sequence[J]. Science, 1998, 282(5388):430-431.
[13]Chang HS, Lin CH, Chen YC, et al.Using siRNA technique to generate transgenic animals with spatiotemporal and conditional gene knockdown[J]. Am J Pathol, 2004, 165(5):1535-1541.
[14]张曼玉, 陈志胜, 计慧琴, 等. 鸡胚胎干细胞分离方法和培养体系的优化[J]. 中国组织工程研究与临床康复, 2011(45):8429-8433.
[15]Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells[J].Science, 2002, 296(5567):550-553.
[16]庄淑珍, 窦忠英, 杨炜峰, 等. ES-D3细胞nanog基因干扰载体的构建及其干涉效果[J]. 农业生物技术学报, 2005(6):754-758.
