N2O和CO2在N+Fe/TiO2(101)面吸附的第一性原理研究
2014-06-27李宗宝
李宗宝,王 霞
(1.铜仁学院物理与电子科学系,固体材料与元器件研究所,贵州铜仁554300;2.铜仁学院生物科学与化学系,贵州铜仁554300)
N2O和CO2在N+Fe/TiO2(101)面吸附的第一性原理研究
李宗宝1,王 霞2
(1.铜仁学院物理与电子科学系,固体材料与元器件研究所,贵州铜仁554300;2.铜仁学院生物科学与化学系,贵州铜仁554300)
基于周期性密度泛函理论,研究了N2O和CO2气体在N/Fe共掺杂锐钛矿TiO2(101)面最稳定结构的吸附,并与其在洁净TiO2(101)面的结果进行了对比.详细比较了三原子气体不同吸附位、不同吸附端在Fe位及N位吸附的吸附能、键长和键角的变化.结果表明:N2O在Fe位吸附较清洁表面强,为化学吸附;CO2在改性表面的吸附较清洁表面弱.
锐钛矿TiO2;密度泛函理论;N/Fe共掺杂;吸附能;三原子分子
0 引言
迄今为止,社会经济的不断发展在给人们带来丰厚物质财富的同时,大量环境污染物的排放对人们生活和健康所带来的威胁日益明显.引起人们广泛关注的是过量排放的温室气体对环境的不良影响.N2O由于在大气中存留时间较长,导致臭氧层损耗较大,是温室效应的主要元凶;CO2作为工业等排放的主要温室气体,其排放量巨大,是气候变暖的主要原因.当前对温室气体的处理方法主要有臭氧处理法、γ射线辐射法、超声波分解法、电化学氧化法和光催化氧化法等[1-5].由于光催化环境友好、过程可控、绿色无污染等优良特性,因此,光催化氧化法受到人们的青睐,由此带来的新型光催化剂的制备与研究亦受到人们的普遍关注.作为众多新型光催化剂中的一种,TiO2因具备制备容易、化学稳定性强、无二次污染和应用广泛等优良特点,成为当前研究的热点.
纯TiO2禁带宽度较大(3.0~3.2eV),只对紫外光有响应,对可见光的利用效率较低(4%).实验发现:通过贵金属沉积,如Ag/TiO2,Pt/TiO2,Sn/TiO2,Au+Pt/TiO2,Au/TiO2等[6-10],可以有效提高光催化活性,并扩大对可见光的利用效率.但是,对贵金属依赖会大大增加其制造成本,较难满足工业化生产的需求,因此必须寻求廉价替代品.实验上,H.Park等证明Fe3+/Fe2+沉积于TiO2表面能够提高其光催化氧化能力[11];N.Nakajima等发现少量Fe吸附于金红石相TiO2(110)面时能诱发材料呈现出非金属性[12],而随着吸附量的增加则表现出金属性;苏碧桃等发现Fe3+因替代Ti4+而使表面显负性[13],从而提高光催化性;我们前期计算结果也证明通过N/Fe表面共掺杂可以有效降低TiO2(101)面禁带宽度并使其表现出半金属性[14],从而成为替代贵金属表面沉积改性的最佳选择.
Wanbayor等详细计算了N2O在TiO2(101)清洁表面及负性表面吸附及光催化氧化过程[15].但未见对于在改性TiO2(101)表面的吸附系统研究.本文在前期工作基础上,研究了N2O和CO2气体在N/Fe共掺杂锐钛矿TiO2(101)面最稳定结构(M—TiO2(101))的吸附,详细对比了N2O和CO2气体在Fe位及N位不同吸附位、不同吸附端吸附时的吸附能以及吸附后的键长和键角.为对不同吸附进行区分,N2O的4种吸附形式分别表示为ONN—Fe/M—TiO2,NNO—Fe/M—TiO2,NNO—N/M—TiO2和ONN—N/M—TiO2,对于CO2则为CO2—Fe/M—TiO2和CO2—N/M—TiO2.其中ONN—Fe/M—TiO2形式中ONN表示N1原子端吸附,Fe表示原子吸附位,其他表示方式与之相同.为有效对比吸附后的稳定性,将上述改性表面吸附与在洁净TiO2(101)表面的吸附结果进行了对比.本文计算所选择的M—TiO2(101)晶体结构如图1所示.
1 计算方法
基于密度泛函理论的平面波超软赝势方法,在周期性边界条件下,采用广义梯度近似下GGA+PBE处理电子间的交换关联能[16].在倒格子空间[17],平面波截断能取Ecut=400eV,Monkhorst-pack特殊K点取为4×4×1.本文所有优化计算均在VASP软件包中进行[18].计算中为防止发生镜像反应,真空层厚度取为12nm.为修正GGA理论在计算能带时存在的缺陷,计算中采用GGA+U[19]方法对态密度进行修正,选参数U=6.3eV和J=1eV.在晶体优化过程中,除底层原子外其余位置原子均开放优化.
为有效获得N2O和CO2气体的真实吸附位,计算了在M—TiO2(101)面上N位和Fe位吸附时的吸附能ΔEads,采用的计算公式为

其中:Eads/slab为吸附后体系总能量;Eslab为吸附前M—TiO2(101)的能量;EGas为吸附气体的能量.根据定义,吸附能为负值,吸附过程是放热过程,则吸附是稳定的;吸附能为正值,吸附过程是吸热过程,则吸附是不稳定的.
2 结果与讨论
2.1 N2O的吸附
在紫外光照射条件下,N2O在TiO2(101)表面的光催化氧化过程见图2.N2O分子含有16个电子,当在TiO2晶体表面进行吸附时,作为施主其总能量减小.即光生空穴促使N2O在TiO2晶体表面进行吸附的同时,光生电子则被周围的受体分子捕获,从而实现表面吸附物的氧化和还原过程.
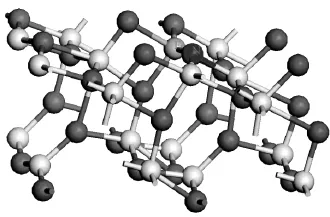
图1 N和Fe共掺杂TiO2(101)面最优化结构
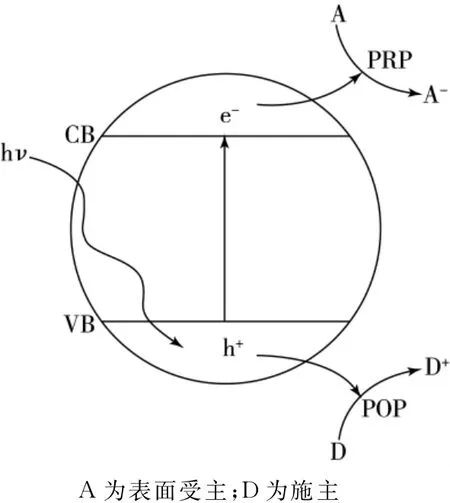
图2 紫外激发条件下TiO2光催化氧化还原过程
理论计算发现[19],N2O气体在纯净TiO2(101)面倾向于Ti5c位吸附,吸附能为-0.17eV,为物理吸附,并表现出稳定的成键趋势,Ti…N1化学键为0.260nm;对于负性表面,N2O的吸附较弱,吸附能仅为-0.01eV.实验和理论证明[20]:表面缺陷的出现对于提高TiO2的光响应及光催化氧化能力具有较大的作用.为有效获得N2O在N和Fe共掺杂改性TiO2(101)面的吸附,本文详细讨论了N1端和O端分别在表面Fe位及N位的吸附.对上述不同吸附进行了优化,并讨论了吸附能和键长的变化.对吸附后的结构进行优化,结果如图3所示.与之相应的吸附能及键长的变化如表1和2所示.为了进行对比,表1和2也列出了前人的计算结果.通过图1可以看出,在M—TiO2(101)面:相同吸附端,N2O在Fe位吸附较N位吸附效果佳,且前者与表面的吸附距离较后者小;相同吸附位时,N1原子端吸附较O原子端吸附更强;NNO—Fe/M—TiO2吸附时N—N—O方向与晶体表面波形出现平行趋势,其他3种吸附N—N—O则垂直于表面.
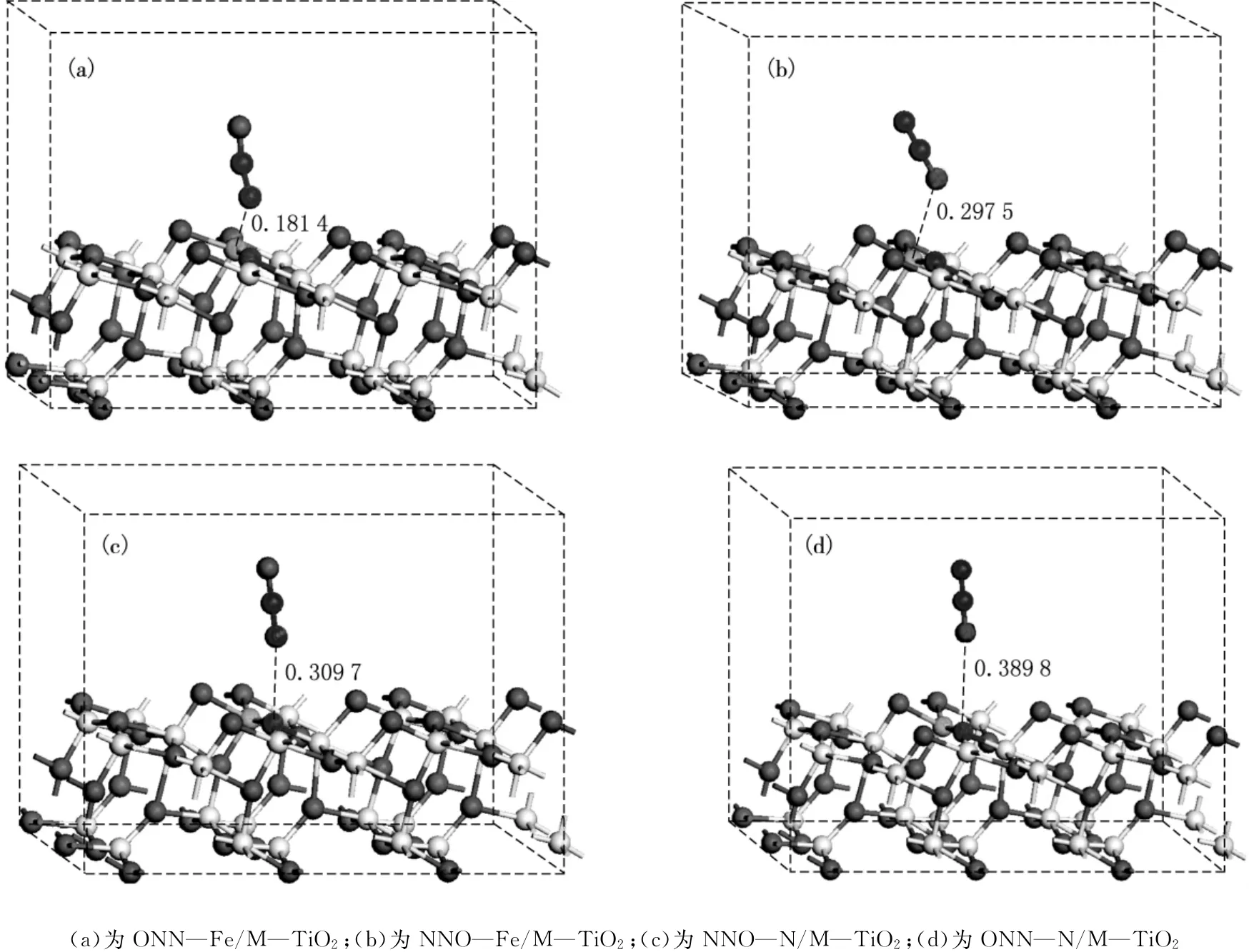
图3 N2O在M—TiO2(101)面N、Fe位吸附的优化结构图
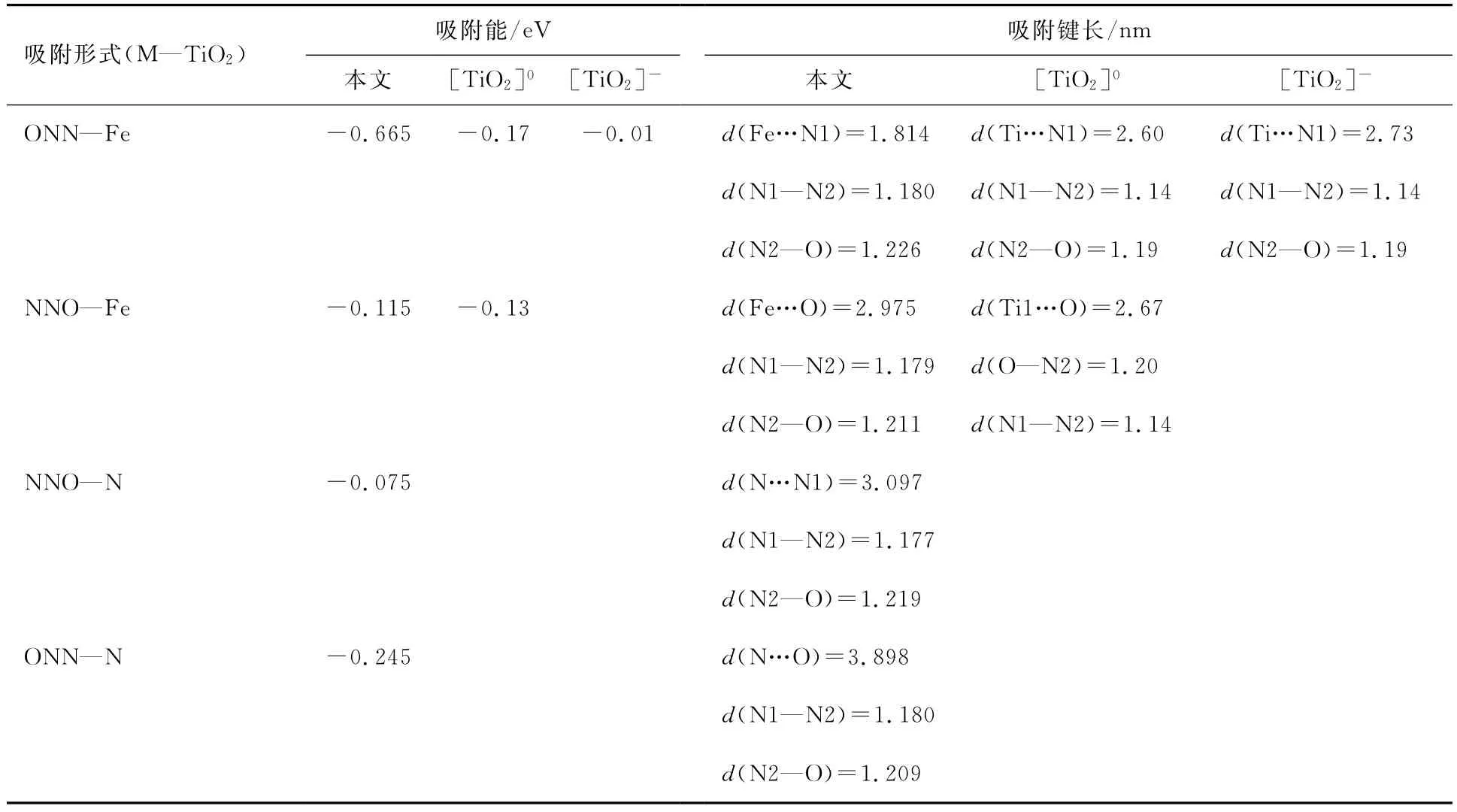
表1 N2O在共掺杂改性TiO2(101)面N和Fe位置吸附的吸附能、键长
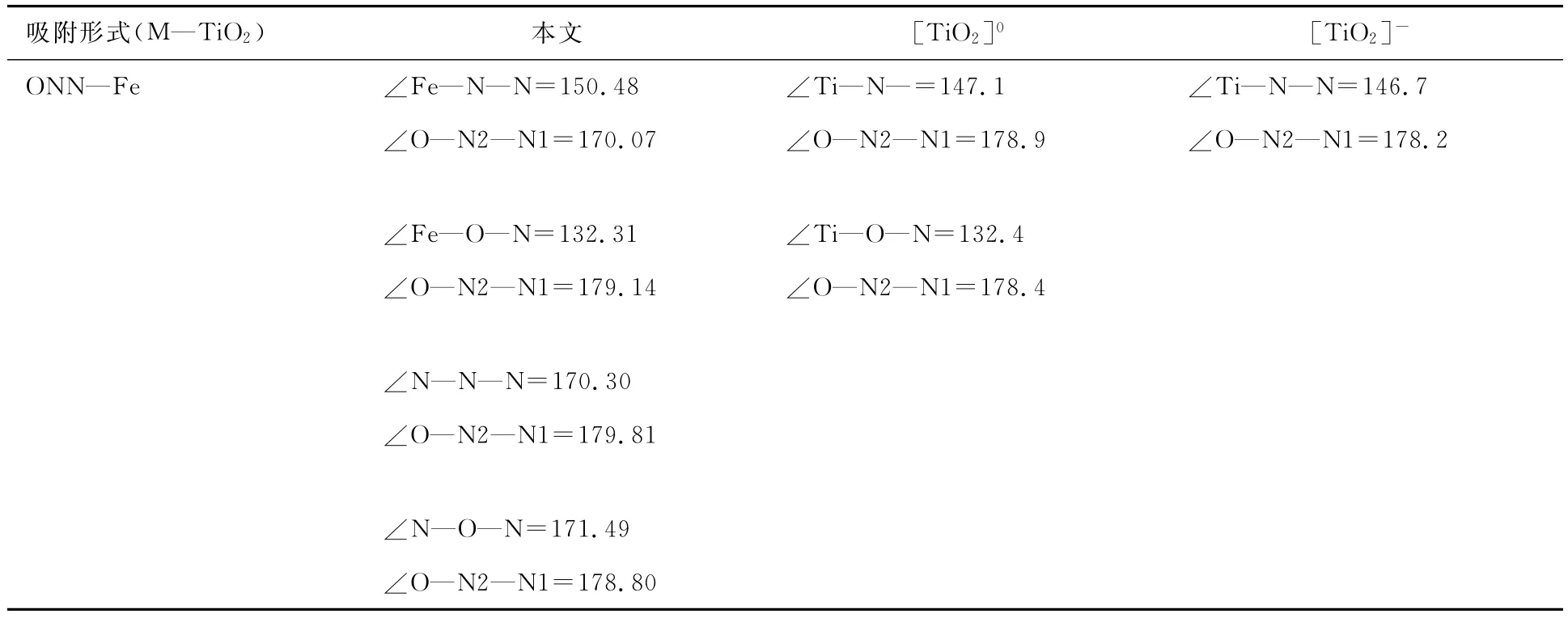
表2 N2O在共掺杂改性TiO2(101)面N和Fe位置吸附的键角(°)
为进一步讨论N2O在M—TiO2平面吸附的变化,讨论了不同位吸附的吸附能、键长和键角,并与清洁表面和负性平面进行了对比.通过表1和2可以看出:(1)N1原子端吸附于Fe位时吸附能最大,为-0.665eV,N1原子端吸附于N位时吸附能次之,为-0.245eV,O原子端吸附时吸附能较小,Fe位吸附为-0.115eV,N位为-0.075eV;(2)4种吸附中,N1—N2—O各键的键长保持变化;(3)N1端吸附于Fe位时,N1…Fe键长最短,为0.181 4nm,N1吸附于N位时N…N1键长最长,为0.389 8nm;(4)N1端吸附于Fe位时,O—N2—N1的键角变化最大,由直线结构变为170.07°,其他三类吸附则几乎保持直线不变.通过上述分析发现,N2O在M—TiO2(101)表面吸附时更倾向于N1原子端吸附于Fe位并形成稳定的N—Fe键.通过与清洁表面和负性表面的对比可以看出:(1)较清洁及负性表面,N2O在M—TiO2(101)表面掺杂Fe位吸附更强;(2)在M—TiO2表面吸附时,O—N2—N1的键长及键角的变化与清洁及负性表面吸附时变化趋势相同.
2.2 CO2的吸附
R.Wanbayor等计算CO2作为三原子气体在洁净TiO2(101)表面的吸附时[21],发现其吸附能较小,为物理吸附.为进一步讨论CO2在M—TiO2(101)面的吸附情况,本文详细计算了CO2在改性表面的吸附能及吸附后的优化结构,如图4和表3所示.为了与洁净表面吸附结果相对比,图4(c)给出了在洁净表面的吸附[21],为了更加深入对比CO2在改性前后(101)表面吸附的不同,表3给出了改性前后表面吸附的吸附能及优化后的结构参数.通过图4和表3可以看出:(1)CO2在M—TiO2(101)面的吸附均为物理吸附;(2)C在Fe位的吸附距离较N位大,在清洁表面的吸附距离最小;(3)三类吸附中,CO2均保持直线型结构;(4)三类吸附中O—C—O线性结构均垂直于TiO2的波形结构.通过表3可以看出,CO2在M—TiO2(101)面Fe位和N位的吸附能均较清洁表面(-0.41eV)低,其大小顺序为E—Fe/M—TiO2(101)<E—N/M—TiO2(101)<E—TiO2(101),表明表面经过改性后对CO2的吸附影响较弱.
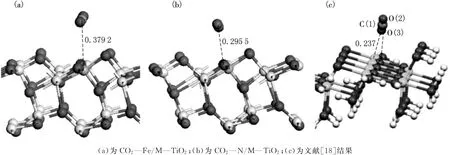
图4 CO2在M—TiO2(101)面N和Fe位吸附的优化结构
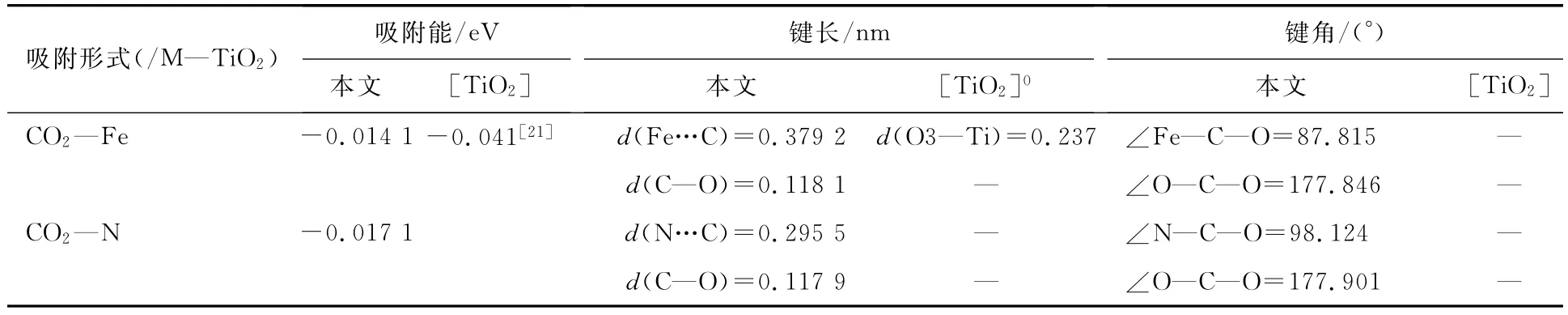
表3 CO2在共掺杂改性TiO2(101)面N和Fe位置吸附的吸附能、键长及键角
通过对N2O和CO2在改性表面的吸附对比可以看出,改性后的M—TiO2(101)表面对N2O的吸附作用较清洁表面的吸附作用强;改性表面对CO2的吸附作用与N2O的作用效果相反.出现该现象的原因可以理解为:TiO2(101)面经过N+Fe共掺杂改性后,其表面出现较纯净表面多余的电荷,该电荷的出现将加强表面与极性分子(如N2O)的吸附作用,并出现成键趋势,形成较强的化学吸附,而对于非极性分子(如CO2)则由于其分子内部饱和键的存在,较难与改性表面电荷发生相互作用,只出现较弱的物理吸附.
3 结论
基于密度泛函理论的第一性原理,计算了极性分子N2O和非极性分子CO2在N+Fe共掺杂锐钛矿TiO2(101)面不同位置的吸附能及吸附优化后的结构.通过对吸附能的比较得出:N2O在Fe位吸附时倾向于N1原子端吸附,并形成稳定的N—Fe键,且较清洁表面的吸附更稳定;CO2吸附与N2O相反,更倾向于在晶体清洁表面进行.通过对比N2O和CO2分子的极性可以得出:N+Fe改性TiO2(101)表面较清洁表面对极性分子气体的吸附作用更强,而对非极性分子则不然.
[1] IKEHATA K,EL-DIN M G.Aqueous pesticide degradation by ozonation and ozone-based advanced oxidation processes:a review(partⅡ)[J].Ozone-sci Eng,2005,27:173-202.
[2] VINODGOPAL K,PELLER J.Hydroxyl radical-mediated advanced oxidation processes for textile dyes:a comparison of the radiolytic and sonolytic degradation of the monoazo dye acid orange 7[J].Res Chem Intermediat,2003,29:307-316
[3] HUNG H M,KANG J W,HOFFMANN M R.The sonolytic destruction of methyl tert-butyl ether present in contaminated groundwater[J].Water Environ Res,2002,74:545-556.
[4] YANG S Y,CHOO Y S,KIM S,et al.Boosting the electrocatalytic activities of SnO2electrodes for remediation of aqueous pollutants by doping with various metals[J].Appl Catal B,2012,111/112:317-325.
[5] GULTEKIN I,INCE N H.Synthetic endocrine disruptors in the environment and water remediation by advanced oxidation processes[J].J Environ Manage,2007,85:816-832.
[6] SUBRAHMANYAM A,BIJU K P,RAJESH P,et al.Surface modification of sol gel TiO2surface with sputtered metallic silver for Sun light photocatalytic activity:Initial studies[J].Sol Energy Mat Sol C,2012,101:241-248.
[7] ZHANG M L,YUAN Z H,NING T,et al.Growth mechanism of Pt modified TiO2thick film[J].Sensor Actuat B-Chem,2013,176:723-728.
[8] ZHANG M Y,HE G Z,DING C C,et al.Mechanism of arsenate(Ⅴ)adsorption on TiO2surfaces[J].Acta Phys-Chim Sin,2009,25:2034-2038.
[9] GONG X Q,SELLONI A,DULUB O,et al.Small Au and Pt clusters at the anatase TiO2(101)surface:behavior at terraces,steps,and surface oxygen vacancies[J].J Am Chem Soc,2007,130:370-381.
[10] YOGI C,KOJIMA K,TAKAI T,et al.Photocatalytic degradation of methylene blue by Au-deposited TiO2film under UV irradiation[J].J Mater Sci,2009,44:821-827.
[11] PARK H,LEE J,CHOI W.Study of special cases where the enhanced photocatalytic activities of Pt/TiO2vanish under low light intensity[J].Catal Today,2006,111:259-265.
[12] NAKAJIMA N,KATO H,OKAZAKI T,et al.Photoemission study of the modification of the electronic structure of transition-metal overlayers on TiO2surfaces:I.Fe on TiO2(110)[J].Surf Sci,2004,561:79-86.
[13] 苏碧桃,孙佳星,胡常林,等.Fe3+掺杂TiO2光催化纤维材料的制备及表征[J].物理化学学报,2009,24(8):1561-1566.
[14] LI Z B,WANG X,JIA L C.Synergistic effects in Fe/N codoped anatase TiO2(101)surface:a theoretical study based on density functional theory calculation[J].Acta Phys Sin,2013,20:203103.
[15] WANBAYOR,DEÁK P,FRAUENHEIM T.First-principles investigation of adsorption of N2O on the anatase TiO2(101)and the CO pre-adsorbed TiO2surfaces[J].Comp Mater Sci,2012,58:24-30.
[16] PERDEW J P,BURKE K,EMZERHOR M.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77:3865-3871.
[17] MONKHORST H J,PACK J D.Special points for Brillouin-zone integrations[J].Phys Rev B,1998,13:5188-5192.
[18] KRESSE G,FURTHERMULLER J.Efficient iterative schemes for ab initio total-energy calculations using aplane-wave basis set[J].Phys Rev B,1996,54:11169-11174.
[19] DUDAREV S L,BOTTON G A,SAVARSOV S Y.Electron-energy-loss spectra and the structural stability of nickel oxide:An LSDA+U study[J].Phys Rev B,1998,57:1505-1510.
[20] WANBAYOR R,RUANGPORNVISUTI V.Adsorption of CO,H2,N2O,NH3and CH4on the anatase TiO2(001)and(101)surfaces and their competitive adsorption predicted by periodic DFT calculations[J].Mater Chem Phys,2012,124:720-725.
[21] WANBAYOR R,RUANGPORNVISUTI V.Adsorption of di-,tri-and polyatomic gases on the anatase TiO2(001)and(101)surfaces and their adsorption abilities[J].J Mol Struct-Chem,2010,952:103-108.
The adsorptions of N2O and CO2on Fe+N/TiO2(101)surface:a theoretical study based on DFT calculation
LI Zong-bao1,WANG Xia2
(1.Department of Physics and Electronic Science &Institute of Solid Materials and Components,Tongren University,Tongren 554300,China;2.Department of Biology Science and Chemistry,Tongren University,Tongren 554300,China)
The adsorptions of N2O and CO2on N and Fe modified anatase TiO2(101)surface are investigated by means of periodic density functional theory.The calculated results are compared with the pure TiO2(101)surface.The adsorption energies,the change of the bond length and angles are calculated with different adsorption forms.The adsorption energy calculations suggest that N2O prefers adsorbed on the N site with the chemisorption while CO2prefers on the pure surface.
anatase TiO2;DFT;N and Fe codoped;adsorption energy;triatomic molecule
O 647 [学科代码] 150·30
A
(责任编辑:石绍庆)
1000-1832(2014)02-0093-06
10.11672/dbsdzk2014-02-019
2013-11-24
贵州省自然科学基金资助项目(黔科合J字LKT[2012]17号);贵州省教育厅自然科学基金重点项目(黔教合KY字[2013]182号).
李宗宝(1982—),男,副教授,主要从事新型功能材料的制备及性能研究.
