警惕造成“血管衰竭”的一大病因—餐后高血糖
2014-06-01井上辉雄
井上辉雄 等
临床综述
警惕造成“血管衰竭”的一大病因—餐后高血糖
井上辉雄 等
餐后高血糖表现为餐后血糖的高峰,这可以引起内皮细胞功能障碍、炎症反应和氧化应激,进而导致动脉粥样硬化的进展和心血管事件的发生。已有资料显示餐后高血糖甚至糖耐量减低即可使动脉粥样硬化和心血管事件更易于发生。有证据显示餐后高血糖可独立预测心血管事件的发生,而空腹高血糖则不能。我们提出“血管衰竭(vascular failure)”的概念,用以概括血管功能障碍的所有表现,包括从危险因素到动 脉粥样硬化性疾病晚期。因此餐后高血糖是造成血管衰竭的一种非常重要的病理生理状态。因此,控制餐后高血糖应该作为预防血管衰竭一个潜在的目标而成为未来临床研究的焦点。
引言
2型糖尿病可使冠状动脉粥样硬化及脑血管疾病的发生风险显著升高。另外,有证据表明,餐后血糖异常状态尤其是餐后高血糖是动脉粥样硬化的独立危险因素。最近的流行病学调查显示,餐后高血糖是心血管疾病的独立危险因素,它对心血管疾病的影响比空腹高血糖更大。
动脉粥样硬化是血管壁对多种原因造成慢性损伤的反应引起的一种进展性疾病,最终导致动脉粥样硬化或纤维斑块的形成。通常认为内皮细胞功能障碍是动脉粥样硬化的起始阶段。除此以外,动脉粥样硬化进程的早期阶段即存在血管壁平滑肌细胞功能障碍、代谢异常,包括炎症反应、氧化应激和神经激素失衡等。我们最近提出“血管衰竭(vascular failure)”的新概念,用以代表上述各种血管异常的整体情况。Schwartz等人曾使用“血管衰竭”一词表示血管重塑反应丧失。与之不同,我们把“血管衰竭”定义为一种血管功能障碍的整体性的综合征,涵盖从危险因素到动脉粥样硬化晚期的动脉狭窄,直至最终导致血管壁钙化,以及斑块破裂或血栓栓塞等严重血管事件(图1)。
餐后高血糖的病理生理过程:血糖高峰引起氧化应激,生成可溶性糖基化终末产物(AGEs)及脂质过氧化物,并作为上游激酶的关键性活化因子,导致内皮细胞功能障碍及炎症基因表达(图2)。餐后高血糖在血管衰竭发病过程中是一种重要的基本功能紊乱,本文回顾餐后高血糖在其中的作用。
最近有证据表明,有症状的心血管疾病患者中将近2/3存在血糖稳态失衡。这些患者绝大部分空腹血糖水平未见升高,而在进食后或口服葡萄糖耐量试验时发现餐后血糖水平升高。葡萄糖不耐受通常由75克葡萄糖耐量试验方可确定。葡萄糖负荷后2小时血糖水平140~200毫克/分升可诊断为糖耐量减低(IGT)或称糖尿病前期,若超过200毫克/分升即可诊断2型糖尿病。

图1 血管衰竭
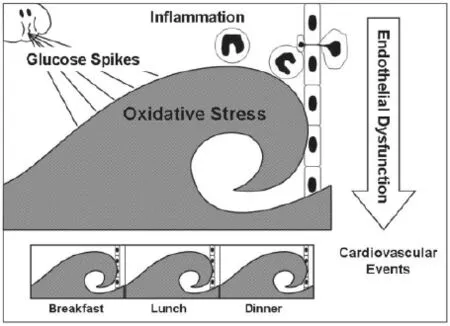
图2 餐后高血糖的病理生理
即使在通过糖化血红蛋白A1c(HbA1c)和空腹血糖水平评估认定血糖控制良好的糖尿病患者中,餐后高血糖也经常发生。人群研究表明,空腹血糖水平低至90毫克/分升仍可能伴随着餐后2小时血糖水平>200毫克/分升。在2型糖尿病的早期阶段,即使空腹血糖与HbA1c在正常范围,餐后高血糖同样会引起微血管并发症以及心肌梗死、卒中等大血管并发症。新近资料显示,即使IGT也可使动脉粥样硬化和心血管事件更易于发生。有证据表明,餐后高血糖可独立预测心血管事件,而空腹高血糖不能。例如,Funagata糖尿病研究(Funagata Diabetes Study)结果表明,与空腹血糖和糖化血红蛋白相比,负荷后1小时或2小时血糖水平能更好的预测心血管风险,而糖尿病流行病学:欧洲糖尿病诊断标准合作分析(Diabetic Epidemiology:Collaborative Analysis of Diagnosis Criteria in Europe,DECODE)显示,负荷后2小时血糖水平与心血管死亡风险间存在直接的连续级配关系。即使在糖耐量正常(负荷后血糖<140毫克/分升)患者中,负荷后血糖水平也与心血管死亡及全因死亡风险相关。负荷后血糖达到80毫克/分升后心血管风险即开始升高,达到140毫克/分升(传统归为IGT或糖尿病前期)时,心血管风险升高58%。
内皮细胞功能障碍是动脉粥样硬化初始阶段的观点现已得到广泛的认同。内皮细胞功能障碍时,血管内皮细胞来源的舒张因子尤其是L-精氨酸在内皮一氧化氮合酶(eNOS)作用下生成的一氧化氮(NO)生成减少,生物利用度降低。NO生物利用度减少导致依赖内皮细胞的血管舒张作用损害,这就是内皮细胞功能障碍的功能性表现。另一方面,它还包含一个特定的“内皮激活”的状态,表现为促炎、增生及血栓前状态,这种状态对动脉粥样化形成的各个阶段均有促进作用。鉴于内皮细胞功能障碍和动脉粥样硬化之间的这种关系,内皮细胞功能状态很可能反映了个体发生动脉粥样硬化性疾病的倾向性。因此,内皮细胞功能障碍可以作为早期血管衰竭的标志。
不仅糖尿病时内皮细胞功能损害,IGT时也是同样。我们观察到与糖耐量正常的受试者相比,IGT患者血流介导的血管舒张(FMD)减弱。血糖迅速升高可影响内皮细胞功能,体内、体外实验均证实高血糖具有这种直接的作用。体外实验中仅通过简单的将标本暴露于高浓度的葡萄糖环境中,就发现乙酰胆碱介导的依赖内皮细胞的血管舒张作用减弱,并表现出浓度依赖性。体内实验同样显示血糖高峰诱导内皮细胞功能障碍,Williams等人利用FMD技术发现,动脉灌注50%右旋葡萄糖引起的急性高血糖,可使健康受试者依赖内皮细胞的血管舒张作用减弱。这个结果表明血糖水平升高是糖尿病相关的内皮细胞功能障碍的一个主要原因。Shige等人也发现2型糖尿病患者进食高脂肪或高蔗糖食物后FMD减弱。上述研究中餐后FMD水平及变化程度均与餐后血糖水平变化显著相关,因此可以确认餐后高血糖是FMD减弱的一个决定性因素。
高血糖的这些作用可能与NO的生成和/或生物利用度减少有关,高血糖诱导的内皮细胞功能障碍可能被精氨酸的生成增加所拮抗。NO的生成与生物利用之间的相互关系尚未明确,尚未明确高血糖是否能减少NO的生成,或者,更有可能的是,NO生成的增加引起它的抑制物超氧阴离子的显著增加,进而导致NO生物利用度的降低。而且已经发现,IGT患者进行OGTT试验时FMD的迅速降低与2小时血糖水平相关。
最近的资料表明,AGEs可以影响内皮细胞功能障碍的进展。AGEs是一组具高度异质性的化合物,最具代表性的是羧甲基赖氨酸(CML)。饮食是外源性AGEs的一个主要来源,食物AGEs含量与食品营养成分含量高度相关,另外也与烹饪的方法、温度及持续时间有关。饮食来源的AGEs大约10%迅速吸收,部分保留在体内,结合并激活AGE受体,进而起到多种病理作用。AGE前体如甲基乙二醛(MG)也可以激活AGE受体,有体内研究显示高血糖与内生MG合成增加相平行。进餐后,吸收和内生的AGEs和MG协同作用,直接清除NO,促进氧化应激,进而降低血管功能。
餐后高血糖与炎症反应
现已公认炎症反应是血管衰竭发病机制中的一个主要因素。长期以来,一直通过炎症标志物如高敏C-反应蛋白(hsCRP)来评价糖尿病时的炎症反应。炎症细胞和介质在炎症过程中起着至关重要的作用。动脉粥样硬化起始及进展时,白细胞如单核细胞和T-淋巴细胞在动脉血管壁内皮浸润,内皮与白细胞之间通过黏附分子调节其相互作用。在各种黏附分子中,细胞间黏附分子-1(ICAM-1)和血管细胞黏附分子-1(VCAM-1)特别引人注意。已经证实血管疾病和糖尿病(不论其是否有血管疾病)时这些分子的循环形式增加。我们发现与健康受试者相比,IGT患者的循环ICAM-1水平已有升高,但VCAM-1未见这种变化(图3)。此外已经证实在正常和糖尿病受试者中,急性高血糖均可以增加循环ICAM-1水平,进而激活动脉粥样硬化进程的起始阶段。现已有证据证实高葡萄糖钳夹或进餐后引起的急性高血糖均可促进血浆白介素-6、肿瘤坏死因子-α及白介素-18生成增加,因此,现已公认动脉粥样硬化是一种炎性疾病,即使在合并糖尿病时也是这样。
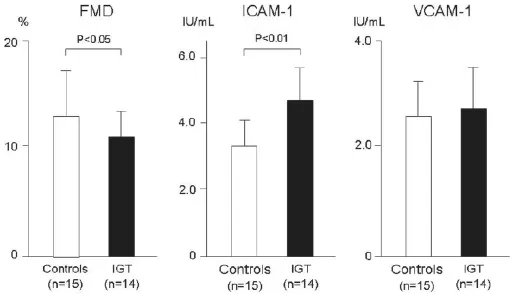
图3 糖耐量减低(IGT)患者与正常糖耐量受试者(对照组)内皮细胞功能与炎症反应标志物
先前的研究主要集中在单核细胞和T-淋巴细胞方面,因为它们是参与动脉粥样硬化的炎症细胞的主要类型,但越来越多的证据表明,中性粒细胞活化在缺血性心血管疾病的炎症进程,特别是在急性炎症应答中,也扮演了重要的角色。此外一项对冠状动脉疾病和脑血管疾病高危患者的大规模队列研究显示,中性粒细胞计数在白细胞分类中是未来心血管事件最好的预测因子。外周中性粒细胞是活性氧最重要的来源。中性粒细胞的激活可显著影响2型糖尿病的氧化应激和炎症反应,继而导致血管病及动脉粥样硬化。β2-整合素Mac-1(CD11b/ CD18)是一种对中性粒细胞及单核细胞紧密黏附于血管内皮细胞起到至关重要的作用的黏附分子。Mac-1与血管内皮表面ICAM-1结合,并通过结合纤维蛋白原或血小板受体如糖蛋白(GP)Ibα、ICAM-2等而与血小板结合。临床研究表明,Mac-1在冠状动脉血管成形术后粘附在损伤血管处的中性粒细胞表面激活和上调。2型糖尿病患者中也发现中性粒细胞表面Mac-1上调。另外有报道体外实验中,激动剂诱导糖尿病患者中性粒细胞表面Mac-1上调增强。此前我们曾对未诊断糖尿病、空腹血糖<126毫克/分升的患者,在口服75克葡萄糖负荷120分钟及以前,使用流式细胞术观察孤立中性粒细胞表面Mac-1的表达情况。结果显示,与基线相比,餐后高血糖(120分钟血糖水平≥200毫克/分升)患者葡萄糖负荷120分钟后,Mac-1甚至在未激活的中性粒细胞表面上调。此外与基线相比,餐后高血糖及IGT(120分钟血糖水平≥140毫克/分升)患者fMLP介导的Mac-1上调在120分钟时显著增强,而这些变化在正常糖耐量(120分钟血糖水平<140毫克/分升)患者中并不明显(图4)。这些结果表明,血糖急性升高可能使餐后高血糖和IGT患者中性粒细胞从非活化形式改变为潜在激活状态。

图4 口服75克葡萄糖负荷后120分钟及口服前β2-整合素Mac-1在孤立中性粒细胞表面的表达
餐后高血糖对氧化应激的影响
氧化应激是细胞的异常氧化还原状态,在血管衰竭尤其是血管内皮细胞功能障碍的发病过程中起到关键的作用。超氧阴离子是氧分子经一价还原形成的,包括黄嘌呤氧化酶、NADH/NADPH氧化酶、脂氧合酶、NOS在内的多种酶类参与这个过程,线粒体是体内超氧阴离子生成的主要场所。超氧阴离子自发的或是经酶催化的歧化反应还原生成过氧化氢,后者经金属(铁或铜)催化转化,生成具有很强毒性的羟基自由基。近来研究显示高血糖介导超氧阴离子在线粒体电子转运链的作用下大量合成超氧阴离子。超氧化物的过量生成造成eNOS与诱导型NOS(iNOS)之间的非耦合状态,导致NO合成增加,进而促进强氧化剂——过氧亚硝基的生成,最终损害DNA。DNA损伤后方可激活细胞核内多聚ADP-核糖聚合酶,继而消耗细胞内的底物NAD+,使糖酵解、电子传递及三磷酸腺苷的生成速度减慢,进而导致磷酸甘油醛脱氢酶活性降低,使二磷酸腺苷核糖化过程减慢。上述这些过程均造成糖尿病血管急性内皮细胞功能障碍,极易引起心血管疾病。现已同时有直接和间接两方面的证据证明,急性高血糖通过氧化应激产物促进IGT受试者心血管疾病的进展。直接证据是基于餐后高血糖氧化应激标志物的作用,如硝基酪氨酸和8-异前列腺素F2α(8-iso-PGF2α)。
氧自由基与一氧化氮反应生成过氧亚硝基,后者一种强氧化剂,可以通过硝基类似物中介使侧链与肽键消除形成羰基从而直接氧化蛋白质、脂类及DNA。过氧亚硝基与酪氨酸残基之间存在亲和力,相互反应生成硝基酪氨酸。一些研究结果表明,高血糖可直接促进硝基酪氨酸的过量生成,例如,猴高血糖时的动脉壁上、健康受试者高血糖钳夹或OGTT时以及糖尿病患者餐后高血糖情况加重时的血浆中均检测到硝基酪氨酸的生成,而且发现硝基酪氨酸的生成依赖于血糖水平。有研究发现大鼠心脏灌注中高血糖伴随着硝基酪氨酸沉积,这似乎与iNOS过度表达使NO与超氧阴离子生成不均衡有关。已经证实硝基酪氨酸的生成可导致健康受试者和灌注心脏冠状动脉的内皮细胞功能障碍。已经证实硝基酪氨酸可直接损伤内皮细胞,因此这种作用并不会令人感到惊奇。
特殊的异前列腺素异构体例如尿液中的8-iso-PGF2α已经公认是氧化应激的标志物。异前列腺素均由自由基介导的花生四烯酸氧化生成,后者广泛分布于细胞膜上,尿液异前列腺素测定能更好的反映整个机体的氧化应激状况。一些研究显示高血糖与8-iso-PGF2α的生成及在尿液中的排泄速度加快有关。据报道,与对应年龄的健康受试者相比,2型糖尿病患者尿液8-iso-PGF2α排泄率显著增加,而且发现血糖水平与尿中8-iso-PGF2α存在显著相关性,提示氧化应激的增加至少可能部分上与糖尿病控制的决定性因素存在相关性。另有体外高血糖条件下培养猪血管平滑肌细胞发现8-iso-PGF2α的合成及释放增加,与上述结果相符合。在最近的一项研究中,Monnier等证实尿液8-iso-PGF2α排泄率与通过平均血糖漂移幅度(MAGE)评估的血糖变异性之间有很强的正相关关系,而且与平均餐后血糖升高间在统计学上具有显著相关性,虽然并非强相关。这些发现表明,与急性餐后血糖高峰相比,急性血糖漂移对氧化应激触发作用的影响更加广泛,因而更应该被纳入血糖紊乱的范畴之内。因此,餐后血糖高峰是“危险的浪潮”,这种认识应该扩展到血糖受间断进食的影响而在平均值上(餐后)下(进餐间隔期间)急性波动。
治疗例证
一些临床试验表明,特定的药物可以降低餐后血糖漂移对总体血糖控制的影响。但到目前为止,尚无评价改善餐后高血糖对糖尿病长期预后影响的前瞻性临床研究。根据餐后血糖对总体血糖控制影响的新近资料,专注于餐后血糖的治疗手段很可能使患者长期受益。传统的糖尿病药物如胰岛素、磺脲类药物等主要降低空腹血糖,而对控制餐后高血糖效果较差。磺脲类药物作用于β细胞钾通道而直接促进胰岛素分泌,但也可能对空腹和餐后血糖均有影响。然而大多数磺脲类药物的药物代谢动力学情况并不针对胰岛素急性分泌,因此他们不能改善胰岛素第一时相分泌异常。另一方面,一些药物的药代动力学及作用机制特别针对餐后高血糖。这样的药物包括格列奈类、α-糖苷酶抑制剂和噻唑烷二酮类药物。
现今非磺脲类促泌剂格列奈类药物包括瑞格列奈、那格列奈和米格列奈,用以恢复第一时相胰岛素分泌。已经证实单剂量的那格列奈通过降低2型糖尿病负荷后血糖而缓解餐后内皮细胞功能障碍。在一项针对糖尿病的随机试验中,磺脲类药物格列本脲降低空腹血糖水平更有效,而瑞格列奈能更大程度的降低餐后血糖水平。两组HbA1c降低的幅度相同,但瑞格列奈可使52%的患者的颈动脉内膜中层厚度得到恢复,而格列本脲组这一比例只有18%,而且动脉粥样硬化的恢复程度与餐后高血糖的降低幅度成正比。
α-糖苷酶抑制剂通过阻断α-糖苷酶而减慢碳水化合物包括淀粉和二糖的分解。糖尿病及葡萄糖耐受不良的患者应用α-糖苷酶抑制剂可使负荷后血糖峰值降低30~70毫克/分升、糖化血红蛋白降低约0.7%,而对空腹血糖水平的影响微乎其微。长期接受阿卡波糖治疗可能通过改善胰岛素敏感性而降低甘油三酯和乳糜微粒的水平。最近的一项研究表明,即使是单次450卡路里的混合饮食就可以立即造成接受饮食治疗的糖尿病患者内皮细胞功能严重的破坏。对同样的受试者预先给予单剂量的阿卡波糖就可以显著降低餐后高血糖,改善进食后的内皮细胞功能障碍。预防非胰岛素依赖型糖尿病(STOP-NIDDM)国际研究共纳入1429例糖耐量减低的受试者,随机分为两组,每日三次随餐服用100毫克阿卡波糖或安慰剂。3.3年后,阿卡波糖使研究的主要终点(新发糖尿病)显著降低25%,并显著降低任何心血管事件发生率达49%、心肌梗死发生率降低91%。阿卡波糖也可延缓颈动脉粥样硬化的进展,减少新发高血压达34%,这些结果说明餐后代谢异常可能参与高血压的发病。一个包括7项长期研究的大型回顾性荟萃分析显示,阿卡波糖可使2型糖尿病心脏事件的风险降低35%,心肌梗死的风险降低64%。此前我们也发现经过3个月的阿卡波糖治疗后FMD增加,hsCRP及VCAM-1水平降低(图5)。这表明阿卡波糖可以改善餐后代谢障碍,改善内皮细胞功能和炎症状态,可能延缓动脉粥样硬化的进展。此外,在最近的一项针对轻症糖尿病的研究中,我们用饮食耐量试验(meal tolerance testing)比较一种新的α-糖苷酶抑制剂——米格列醇与格列奈类——米格列奈对餐后血糖和胰岛素代谢的影响,同时评估其他的葡萄糖代谢相关指标、动脉粥样硬化指标以及肾功能情况。结果表明,尽管两种药物引起的餐后胰岛素分泌模式不同,服用3个月后餐后高血糖的改善情况相近(图6A),米格列醇组1,5-脱水山梨醇水平显著高于米格列奈组;米格列醇组稳态模型胰岛素抵抗指数和尿白蛋白排泄率显著降低,米格列奈组则不明显;米格列醇组血清胱抑素C水平无改变,米格列奈组则升高;米格列醇组hsCRP水平显著降低(图6B)、血清脂联素水平显著升高,而米格列奈组则没有显著改变。上述结果表明米格列醇有抗炎和肾脏保护作用,这可能与改善胰岛素抵抗有关。

图5 阿卡波糖对轻症糖尿病患者血管内皮细胞功能和炎症状态的影响
噻唑烷二酮即过氧化物酶体增殖物激活受体γ(PPAR-γ)激动剂类药物如曲格列酮、罗格列酮、吡格列酮等,可改善胰岛素敏感性,而对胰岛素分泌的影响通常可以不予考虑。然而经由曲格列酮证实,这类化合物应用于糖耐量减低受试者时,可以促进葡萄糖刺激引起的胰岛素分泌,改善餐后高血糖。PPAR-γ激动剂显示出抗炎及通过调节细胞氧化还原状态改善内皮细胞功能的作用。现已证实噻唑烷二酮类药物能降低hsCRP等炎症标志物水平,提高肱动脉的FMD。一项纳入5238名2型糖尿病合并大血管疾病患者的大样本的随机临床试验证实,与安慰剂相比,吡格列酮与降糖药物及其他药物联合使用后可减少心血管不良转归。也有报道与格列美脲相比,吡格列酮可延缓颈动脉内膜中层厚度进展,及通过血管内超声成像评估的冠状动脉粥样硬化的进展。
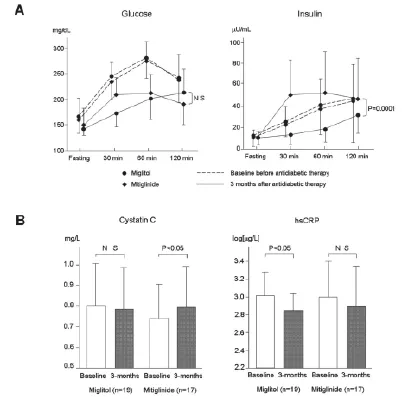
图6 米格列醇与米格列奈对轻症糖尿病患者饮食耐量试验中餐后血糖及胰岛素代谢效用对比
最近一些能带来临床获益的新型降糖药被批准上市,包括注射用药胰高血糖素样肽-1(GLP-1)受体激动剂及口服药物二肽基肽酶-4(DPP-4)抑制剂。GLP-1受体激动剂如艾塞那肽能促进营养物质引起的胰岛素分泌,减少胰高血糖素的不当分泌,同时延缓胃排空,控制食欲。这些药物在坚持减肥时发生低血糖的风险很低。DPP-4抑制剂沙格列汀及维格列汀,对体重通常无影响,胃肠道副作用不如GLP-1受体激动剂严重。无论是单独应用还是与格列本脲或吡格列酮联合应用,此类药物表现出良好的耐受性,而且不发生低血糖;与格列本脲或吡格列酮联合应用可改善2型糖尿病患者的餐后血糖,而对这些药物的药代动力学无显著影响。
结论
餐后高血糖期间,血糖的高峰可导致内皮细胞功能障碍、炎症反应及氧化应激,进而导致动脉粥样硬化的进展及心血管事件的发生。流行病学与发病机制研究的资料均表明,餐后高血糖甚至IGT即可影响动脉粥样硬化性疾病的发生发展,有初步证据表明控制餐后高血糖可以减少IGT人群心肌梗死的发病率。因此,餐后高血糖是导致血管衰竭的一种极为重要的病理生理状态。相应地,控制餐后高血糖应该作为临床随访时预防血管衰竭的潜在目标。
(林乐乙 编译辽宁中医药大学附属第二医院)
10.3969/j.issn.1672-7851.2014.06.008
日本佐贺大学医学院心血管与肾脏学系
