一种基于聚酰胺-胺型树枝状聚合物富集糖肽的新策略
2014-05-04邓珊珊曹琦琛马成白海红任晓君应万涛蔡耘
邓珊珊,曹琦琛,马成,白海红,任晓君,应万涛,蔡耘
1.安徽医科大学,安徽 合肥 230032;2.蛋白质组学国家重点实验室,北京蛋白质组研究中心,军事医学科学院 放射与辐射医学研究所,北京 102206;3.辽宁中医药大学 附属医院,辽宁 沈阳 110032
蛋白质糖基化是最常见的翻译后修饰之一[1]。它不仅在蛋白质的稳定性、溶解性、折叠和功能方面起着关键作用,还在细胞内参与蛋白质的转运、定位和分子识别[2-3]。此外,异常糖基化也被证明和癌症等疾病相关[4-5],许多糖蛋白可以作为临床生物标志物和治疗靶点[6]。因此,糖蛋白和糖肽的分离、发现和鉴定变得越来越重要,它可以帮助我们更好地理解许多生物过程,并找到一些新的潜在的诊断标志物和治疗靶点。以生物质谱技术为主的规模化糖肽定性与定量分析,已成为当前蛋白质翻译后修饰研究的一个重要内容。由于糖肽仅占所有酶解后肽段的小部分(2%~5%)[7],其质谱响应很容易被高丰度非糖肽抑制。另外,由于糖链的微观不均一性,一个糖基化位点上的糖链类型可能多达几十种,进一步降低了糖肽的相对量而使得它们很难被检测到。因此,针对糖肽的特异性富集就显得至关重要。目前,最常用的糖肽富集手段是凝集素亲和[8-9]。不同的凝集素可以特异性地识别不同的糖型,如伴刀豆凝集素ConA对于高甘露糖型和杂合型N-糖都显示出了很好的亲和性。除凝集素亲和富集外,其他手段如分子排阻[10]、亲水色谱[11-12]、酰肼富集[13-14]也都广泛应用于糖肽的富集。但是,各种富集手段都存在一定的缺点。例如,凝集素不能针对所有糖型富集;亲水富集会带来假阳性和假阴性的鉴定结果;肼化学富集法操作繁琐,特异性不强。这些都使得糖蛋白组学研究的进展相对落后于磷酸化、泛素化等翻译后修饰。
树枝状聚合物由于具有高度支化的三维球状立体结构和众多的末端基团,显示出与相应线型分子截然不同的性质[15],如低黏度、良好的溶解性和大量可修饰的末端官能团等,使其具有广泛的应用前景,而且合成相对简单、成本低,因此成为近年来聚合物科学领域的研究热点[16-17]。聚酰胺-胺[poly(amido⁃amine),PAMAM]型树枝状聚合物是其中较典型的、也是当前研究和应用较多的一类[18]。由于树枝状聚合物具有大量端基、分子间无缠结、低黏度和优异的流动性质,并且其表面官能团随相对分子质量和分子尺寸递增,近年来已有很多将树枝状聚合物应用到蛋白质组学研究中的例子[19-21]。在此,我们首次尝试把PAMAM固定于溴化氰活化的琼脂糖凝胶表面,并将之用于糖肽的选择性分离。
1 材料和方法
1.1 材料
5~6周的雄性C57小鼠购自军事医学科学院实验动物中心;第6代PAMAM型树枝状聚合物(PAMAM-G6,10%)、第4代PAMAM型树枝状聚合物(PAMAM-G4,10%)购自山东威海晨源新材料有限公司;溴化氰琼脂糖凝胶(CNBr-activated Sepha⁃rose 4B)、牛血清白蛋白(BSA)、马心肌蛋白(MYO)、牛胎球蛋白(fetuin)、N-糖酰胺酶 F(PNGase F)、α-氰基-4-羟基肉桂酸(CHCA,95%)、三氟乙酸(TFA,99%)、氰基硼氢化钠(NaCNBH3)、高碘酸钠(NaIO4)、尿素、十二烷基硫酸钠(SDS)及己二酸二酰肼琼脂糖(adipic acid dihydrazide-Agarose)均购自Sigma公司;甲酸(FA)购自Acros公司;色谱级乙腈(ACN)、甲醇(CH3OH)购自J.T.Baker公司;1,4-二硫苏糖醇(DTT,98%)、碘乙酰胺(IAA,98%)、测序级胰蛋白酶(15 276 U/mg)购自Promega公司;Amicon Ultra-0.5、30 kD超滤管购自Millipore公司;膜离心吸附柱购自Thermo公司;Sep-Pak C18固相萃取小柱购自Waters公司;所有实验用水由Milli-Q纯水系统提供;其他试剂均为国产分析纯产品。
基质辅助激光解吸附飞行时间质谱(MALDITOF-TOF/MS,AB Sciex公司);Agilent 1100毛细管高效液相色谱-电喷雾-线性离子阱-傅里叶变换离子回旋共振质谱仪(nanoLC-ESI-LTQ-FT,Thermo Finnigan公司);Milli-Q纯水系统(Millipore公司);PB-10型pH计、BP211d分析天平(Sartorius公司);DH-Ⅱ旋转混合器、HS-3垂直混合仪(宁波新芝生物科技股份有限公司);QL-901型涡旋混合仪(其林贝尔仪器制造有限公司);NanoDrop 2000c超微量紫外分光光度计(Thermo Fisher Scientific公司);3K30型高速离心机(Sigma公司);电热恒温水浴箱(北京医疗设备厂)。
1.2 合成固定化的树枝状聚合物材料
取400 μL 1 mmol/L HCl活化60 mg溴化氢琼脂糖凝胶15 min,转至膜离心吸附柱内,用1 mmol/L HCl、结 合 溶 液(0.5 mol/L NaCl和 0.1 mol/L CH3COONa,pH8.3)各洗涤3次(每次200 μL),均分成2份,分别加入50 μL PAMAM-G6冻干粉和50 μL PAMAM-G4冻干粉,加入结合溶液400 μL,室温结合过夜,然后用200 μL结合溶液离心清洗5次,加入 0.1 mol/L 封闭溶液(0.1 mol/L Tris-HCl,pH8.0)常 温 封 闭 2 h,用 清 洗 溶 液 1(0.1 mol/L CH3COONa 和 0.5 mol/L NaCl,pH4.0)、清洗溶液 2(0.1 mol/L Tris-HCl和0.5 mol/L NaCl,pH8.0)各洗4次(每次 200 μL),加入 80%ACN/0.1%TFA 至PAMAM终浓度为2 μg/μL,储存于4℃冰箱。
1.3 蛋白酶切
1.3.1 标准蛋白BSA和牛胎球蛋白酶切 将标准蛋白样品溶解于 50 μL 50 mmol/L NH4HCO3,加入0.5 μL 1 mol/L DTT,蛋白终浓度为 10 mmol/L,95℃变性10 min;加入2.5 μL 1 mol/L IAA,室温暗处放置45 min烷基化,蛋白终浓度为50 mmol/L;加入 1 μL 1 mol/L DTT,封 闭 剩 余 的 IAA;加 入NH4HCO3至蛋白终浓度为2 μg/μL;按蛋白∶酶为1∶50的比例加入胰蛋白酶,37℃水浴酶切过夜;样品存放于4℃冰箱待用。
1.3.2 鼠脑组织蛋白提取及酶切 C57小鼠于实验前16 h禁食,自由饮水;断颈处死后迅速取出鼠脑,浸入冰冷的PBS缓冲液中,洗净残血,去除结缔组织,称重;将脑组织剪碎,加入5倍体积的裂解液(8 mol/L 尿素,4%SDS,0.1 mol/L Tris-HCl,0.1 mol/L DTT,pH7.6),并在研钵内匀浆10次,达到充分碎裂细胞的目的;将样品加入15 mL离心管中,在4℃条件下混匀,15 min后进行超声波提取及离心;超声波仪设置为脉冲工作模式,即工作1 s、间隔1 s,总时间为30 s,循环8次;4℃下混悬30 min,使蛋白充分溶解,最后在10℃下以40 000 r/min离心1 h,取上清液(含有胞浆蛋白和部分亚细胞结构中的膜蛋白)用NanoDrop 2000c测量蛋白浓度后备用。
参照过滤-辅助的样品制备(FASP)[22]方法酶切鼠脑蛋白:取50 μL鼠脑蛋白提取液,加入50 μL 100 mmol/L DTT[用 4%SDS、100 mmol/L Tris/HCl(pH7.6)溶解],95℃变性5 min,转移至30 kD超滤管中,加入200 μL尿素,4℃、14 000 r/min离心15 min,重复1次;加入100 μL 50 mmol/L IAA溶液,550 r/min振荡1 min,室温下暗处烷基化20 min,14 000 r/min离心15 min;加入200 μL尿素,同样转速离心15 min,置换多余的IAA;加入100 μL 50 mmol/L NH4HCO3,14 000 r/min离心10 min,重复2次,弃去滤液;最后加NH4HCO3至蛋白终浓度为2 μg/μL,按蛋白∶酶为1∶50的比例加入胰蛋白酶,37℃烘箱过夜酶切,酶切结束后,14 000 r/min离心10 min;继续加入 100 μL NH4HCO3,14 000 r/min离心10 min;合并滤液,存于4℃冰箱待用。
1.4 糖肽富集
1.4.1 肽段脱盐 用Sep-Pak C18固相萃取小柱对富集前的肽段脱盐。先用1 mL ACN活化小柱,再用1 mL 0.1%TFA溶液平衡小柱,然后将酶切样品溶液(用TFA调整pH值为3)加载于脱盐柱上,并用1 mL 0.1%TFA 溶液 冲洗 1 次 ,最后 用 600 μL 80%ACN/0.1%TFA溶液洗脱肽段,取5 μL洗脱溶液置热干机中热干,等体积水溶后用NanoDrop 2000c定量。
1.4.2 糖肽富集 加入终浓度为10 mmol/L NaIO4至脱盐后的肽段溶液,4℃避光氧化l h,使其将糖单元上的邻二羟基氧化为醛基;向氧化后的溶液中加入0.1%TFA降低ACN浓度,脱盐后溶于400 μL 80%ACN/0.1%TFA;加入一定量的固定化的材料后,于垂直混合仪上混合过夜,取出,稀释至ACN浓度低于10%,转入30 kD超滤管,14 000 r/min离心30 min,用合适的清洗溶剂去除非特异性吸附,直至滤液质谱信号低于50。
1.4.3 糖链切除 按照酶/蛋白为1 U/10 μg的比例将PNGase F加入富集得到的糖肽中,常温酶切3 h,将N-糖基化肽段从聚合物上释放,离心收集,进行质谱分析鉴定。
1.5 质谱采集方法
1.5.1 MALDI-TOF-TOF/MS分析 样品与基质溶液(CHCA溶于50%ACN/0.1%TFA,5 g/L)以1∶1的体积比混合均匀,取0.8 μL点靶,用马心肌红蛋白的胰蛋白酶酶切肽段作为标准物对仪器进行外标校正,相对标准偏差<10 ppm,进行质谱分析。采用正离子反射检测模式进行一级质谱分析。一级质谱数据采集采用MS-1kv反射模式,加速电压20 kV,扫描范围m/z为1000~4000,激光能量4500,每张谱图由1750个亚谱累加获得。
1.5.2 HPLC-ESI-LTQ-FT MS 分析 高效液相色谱采用Agilent 1100毛细管液相色谱仪,自动进样器上样。通过该色谱系统将样品组分进行色谱分离后,由ESI离子源进入质谱进行分析。液相色谱流动 相 A 为 2%ACN、0.1%FA,流 动 相 B 为 80%ACN、0.1%FA。洗脱梯度:0~90 min,6%~40%B;90~105 min,40%~100%B;105~120 min,100%B;用100%A平衡30 min后进行下一次分离。上样量25 μL,流动相流速300 nL/min。质谱选用正离子模式采集数据,扫描范围设置为375.0~1500.0(m/z),采集时间为110 min。选取一级质谱中10个信号最强的离子进行二级分析,其碰撞能量为35 V,采用动态排除功能(dynamic exclusion),排除时间30 s。
1.6 数据库检索与分析
MALDI-TOF-TOF/MS仪器控制软件为4800 Explorer software。ESI-LTQ-FT MS质谱谱图数据检索均使用pFind软件;数据库为UniProtKB mouse(2013-04-01更新),胰蛋白酶为蛋白质水解酶,且最多允许2个漏切位点;半胱氨酸烷基化设置为固定修饰;将甲硫氨酸氧化设置为可变修饰;母离子质量偏差(mass tolerance)为20 ppm;碎片离子设置为0.8 Da。搜索的数据结果用PEAKS软件合并计算,软件参数设置:假阳性率(FDR)≤1%,Deltacn=0.1。糖基化位点通过2个规则确定:①天冬氨酸残基有0.9840 Da质量位移;②天冬氨酸残基必须在N-XS/T/C的结构域中。
2 结果和讨论
2.1 糖肽富集策略的建立及材料表征
本研究目的在于利用PAMAM大量末端氨基来开发一种糖肽富集新材料。首先利用亲和加成反应,将大量N端的树枝状聚合物固定到溴化氰活化的琼脂糖凝胶上,单糖结构中的顺式邻位羟基可以被高碘酸氧化产生醛基,醛基可以与氨基形成希夫碱,高密度的氨基有利于提高反应效率,从而实现糖肽的高效富集。用膜离心吸附柱阻截结合糖肽的聚合物,经PNGase F处理从固相载体上释放糖肽,并采用质谱对糖肽进行序列鉴定和位点确认。由于希夫碱的生成反应是可逆反应,为使反应向正方向进行,提高反应转化率,通常加入一定的还原剂,或通过优化反应条件,如反应时间、温度、pH值和溶剂系统及反应试剂的比例来实现。实验中,我们首先对该聚合物富集条件进行优化,并利用优化后的方法,开展复杂体系中糖肽的特异性富集和鉴定试验。
2.2 材料表征结果
琼脂糖凝胶经第4代和第6代树枝状聚合物修饰前、后的凝胶扫描电镜结果如图1,用放大200倍的扫描电镜能看出凝胶颗粒表面光洁度已有不同,放大1000倍后发现修饰后的颗粒粒径增大了0.1 mm,颗粒表面也凹凸不平,证明材料被成功地固定到固相载体上。进一步以不同放大倍数的透射电镜验证,颗粒修饰前呈均一阴影,而修饰后有很多不均匀的黑点,材料表面凹凸不平,说明不同代数的树枝状聚合物材料都被成功地固定化。
2.3 富集条件的优化
2.3.1 还原剂的考察 PNGase F酶切是最普遍的N-糖链释放方法,几乎可以水解糖肽和糖蛋白中所有的N-糖链,它可以切割分离与糖肽/糖蛋白天冬酰胺残基直接相连的N-乙酰葡萄糖胺(GlcNAc),使天冬酰胺(Asn)转变为天冬氨酸(Asp),产生0.9847的质量变化。对于氨基和醛基的反应,一方面反应完全程度随反应温度升高而不断提高,另一方面当温度大于50℃则易导致PAMAM环化成己内酰胺,造成结构缺陷[23]。因此,所有反应要在低温(25℃~50℃)下进行。考虑到PNGase F的活性,所有实验在室温下进行。富集条件的考察均由PAMAM-G6对标准蛋白牛胎球蛋白的富集结果确定。
由于醛基与氨基之间为可逆反应,温度并不是影响此反应完全程度的关键,该反应的核心问题是使反应不断向正向进行,提高产物转化率。牛胎球蛋白是一个研究较多的糖蛋白,它的胰酶酶切肽段包括3个N-连接和至少3个O-连接糖基化肽段[24]。图2A为30 μg牛胎球蛋白溶于100 μL NH4HCO3经PNGase F酶切得到的质谱图,只能检测到1个糖肽信号(m/z 1741.7),加入 90 μg PAMAM-G6 富集后,糖肽信号峰1741.7有了明显提高(图2B),但仍然不能检测到另外2条糖肽信号,而在偶合反应中加入还原剂NaCNBH3后可以检测到3条糖肽峰(图2C),分别是 m/z 1741.7、m/z 3018.4 和 m/z 3673.7,所以还原剂可以促进反应不断向正向进行。

图1 溴化氰活化的琼脂糖凝胶修饰前后扫描电镜结果A:修饰前;B:经PAMAM-G4修饰后;C:经PAMAM-G6修饰后

图2 牛胎球蛋白酶切肽段在糖肽富集前后的质谱图A:未富集;B:未加还原剂富集;C:加入还原剂富集;N#表示N-连接糖肽序列中的天冬氨酸残基
2.3.2 反应体系酸度的考察 根据文献报道,醛基和氨基在二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)和ACN中反应专一,杂质峰少,因甲醇和四氢呋喃(THF)毒性较大,所以我们选择ACN作为反应溶剂。对于氨基试剂,在碱性催化剂下的反应转化率很低,在酸性催化剂存在下反应转化率较高,实验选取30 μg牛胎球蛋白肽段加入90 μg凝胶材料,考察在不同酸度条件下的富集情况,结果见表1。牛胎球蛋白酶切后产生3条糖肽,当结合溶液pH值不断降低(从3.08到2.18)时,酸度提高,反应转化率增加,从只能检测到1条糖肽到3条糖肽均能被检出;当溶液pH值继续降低(从2.18到0.45)时,质谱检测结果中非特异性吸附的肽段明显增加,这些干扰造成低丰度糖肽被抑制,只能检测到高丰度的糖肽 1741.7。结果表明,以 80%ACN/0.1%TFA(pH2.18)作为结合溶液时,可以特异地富集3条糖肽,最终选择此反应体系。
2.3.3 清洗条件的考察 低代的树枝状聚合物分子具有扩展的或“开放式”结构,随着代数增加,末端基团变得很拥挤,导致了树枝状聚合物采取致密的球状结构,内部具有广阔的空腔,材料在与糖肽结合过程中除了共价结合糖肽分子以外,也可能会非共价包裹一些非糖肽分子,必须采用合适的洗脱溶剂才能有效去除非特异性吸附的非糖肽。我们考察了不同清洗溶剂的清洗情况,依次用500 μL的1.5 mol/L NaCl、8 mol/L 尿 素 、60%CH3OH、50 mmol/L NH4HCO3各洗脱1次,收集每次洗脱得到的溶液,脱盐处理后进行质谱分析,在给定质谱能量下质谱检测信号低于50,我们认为非特异性结合的肽段被去除干净。
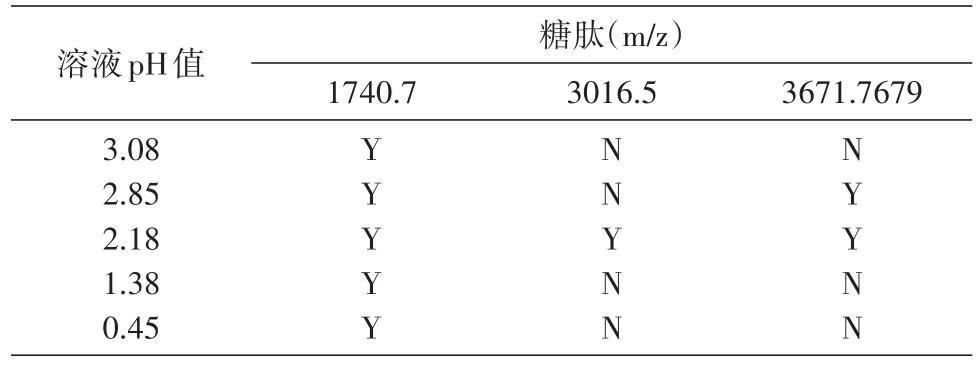
表1 牛胎球蛋白胰蛋白酶酶切肽段在不同pH值结合溶液中富集获得糖肽的结果
2.3.4 反 应 试 剂 比 例 的 考 察 取 不 同 量 的PAMAM-G6对等量的牛胎球蛋白肽段(30 μg)进行富集(质量比分别为0.5∶1、2.5∶1、5∶1、7.5∶1、10∶1),MALDI-TOF-TOF/MS 分析表明糖肽m/z 1741.7峰的强度随PAMAM-G6的用量发生变化(图4),PAMAM-G6富集蛋白的最优比为5∶1,此时基峰强度不再增加,富集效果也达到最好。
2.4 样品复杂性对富集效果的影响
在优化好相关富集条件后,我们探索PAMAMG6针对糖肽富集的选择性,以保证聚合物材料能够从一个较复杂的体系中选择性地分离富集糖肽。将糖蛋白牛胎球蛋白和非糖蛋白BSA的胰蛋白酶酶解产物按一定比例(1∶50,质量比)混合后进行富集,富集前、后的质谱图如图5。可以看到,用PAMAM-G6进行富集前只能检测到大量强度很高的非糖肽峰,经过聚合物材料富集之后,糖肽峰被保留,非糖肽峰减弱,说明材料具备很好的糖肽选择性。
2.5 鼠脑裂解液糖肽富集
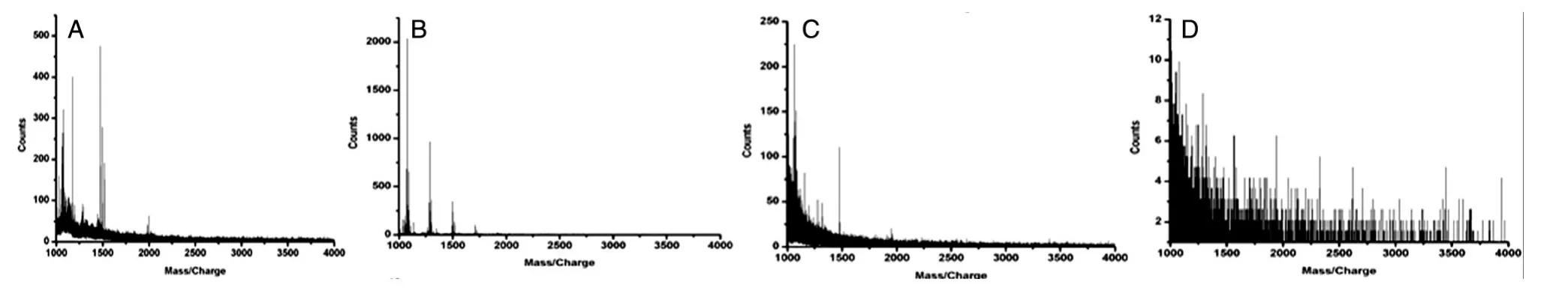
图3 不同清洗溶剂质谱图A:1.5 mol/L NaCl;B:8 mol/L尿素;C:60%CH3OH;D:50 mmol/L NH4HCO3

图4 不同PAMAM-G6用量条件下的糖肽回收情况
通过以上标准糖蛋白考察实验,我们初步建立了优化的PAMAM-G6富集糖肽的新方法:选择反应体系为80%ACN/0.1%TFA,并在结合过程中加入还原剂,材料过量5倍,以 1.5 mol/L NaCl、8 mol/L尿素、60%CH3OH、50 mmol/L NH4HCO3为清洗溶液。我们将这一优化的富集条件应用于200 μg鼠脑全蛋白裂解液液中糖肽的富集,同时采用PAMAM-G4和PAMAM-G6按同样的操作流程富集,作为对比,取市售酰肼化琼脂糖凝胶50 μL用于同量鼠脑全蛋白裂解液中糖肽的富集。利用PEAKS软件搜索质谱raw文件,手动筛选出打分高于15的肽段,得到的结果按照1.6中的规则处理。
结果表明,鼠脑全蛋白裂解液在富集前糖肽占全部肽段比例不到1%,经过商业化酰肼材料富集后鉴定到的糖肽比例增加到14%,说明商业化酰肼材料可以提高糖肽的富集效率。经PAMAM-G4富集后,从鼠脑198条肽段中鉴定出56条非冗余糖肽,占总肽段数目的28%;经PAMAM-G6富集后,从鼠脑632条肽段中鉴定出204条非冗余糖肽,占总肽段数目的32%。很明显,不同代数聚合物对糖肽的选择性均高于商业化酰肼材料。从图6可知,等量的鼠脑全蛋白裂解液经PAMAM-G4富集后鉴定到56条非冗余糖肽,经PAMAM-G6富集后鉴定到204条非冗余糖肽,PAMAM-G4鉴定到的糖肽数目低于PAMAM-G6。我们推测这主要是因PAMAM独特的结构特征所致,1个分子PAMAM-G6含末端氨基256个,而1个分子PAMAM-G4含末端氨基64个,前者的末端官能基团数目是后者的6倍,这种差异导致后者的糖肽结合数目大于前者,富集效率也得到了提高。

图5 牛胎球蛋白和BSA肽段混合物经PAMAM-G6富集前(A)、后(B)的质谱图N#:N-连接糖肽序列中的天冬氨酸残基
2.6 结论

图6 鼠脑糖肽鉴定结果的相关性
目前,有关树枝状聚合物的研究不仅集中在揭示其独特的性质和性能方面,而且逐步转移到功能化和应用上,其应用研究是一个新兴的、很有吸引力的领域。在本研究中,我们首次将不同代数的树枝状聚合物材料PAMAM固定化于溴化氰活化的琼脂糖凝胶表面,并将之用于糖肽富集,采用优化的方法,在鼠脑中富集并鉴定到218条非冗余糖肽,无论是富集糖肽的特异性还是鉴定到的糖肽总数,均较商业化材料显示出更好的效果。我们推测,基于树枝状聚合物的新材料的开发,包括将其固化于纳米磁珠材料、选择更新代数的聚合物和不同的表面反应基团等,将有望更好地实现对糖肽的针对性富集。
[1] Hägglund P,Bunkenborg J,Elortza F,et al.A new strategy for identification of N-glycosylated proteins and unambiguous assignmentoftheirglycosylation sitesusingHILIC enrich⁃ment and partial deglycosylation[J].Proteome Res,2004,3(3):566.
[2] Kazuaki O,Marth J D.Glycosylation in cellular mechanisms of health and disease[J].Cell,2006,126(5):855-867.
[3] Hann S R.Role of post-translational modifications in regulat⁃ing c-Myc proteolysis,transcriptional activity and biological function[J].Semin Cancer Biol,2006,16:288-302.
[4] Slawson C,Hart G W.O-GlcNAc signalling:implications for cancer cell biology[J].Nat Rev Cancer,2011,11,673-684.
[5] Leroy J G.Congenital disorders of N-glycosylation including diseases ssociated with O-as well as N-glycosylation defects[J].Pediatr Res,2006,60(6):643-656.
[6] Freeze H H,Aebi M.Altered glycan structures:the molecu⁃larbasis ofcongenitaldisorders ofglycosylation[J].Curr Opin Struct Biol,2005,15(5):490-498.
[7] Alvarez-Manilla G,Atwood J 3rd,Guo Y,et al.Tools for gly⁃coproteomic analysis:size exclusion chromatography facilitates identification of tryptic glycopeptides with N-linked glycosyl⁃ation sites[J].J Proteome Res,2006,5(3):701-708.
[8] Hiroyuki K,Haruna S,Yoshio Y.Lectin affinity capture,iso⁃tope coded tagging and mass[J].Nat Biotechnol,2003,21(6):667-672.
[9] Dai Z,Zhou J,Qiu S J,et al.Lectin-based glycoproteomics to explore and analyze hepatocellular carcinoma-related glyco⁃protein markers[J].Electrophoresis,2009,30(17):2957-2966.
[10]Qin H,Zhao L,Li R,et al.Size-selective enrichment of N-linked glycans using highly ordered mesoporous carbon materi⁃al and detection by MALDI-TOF MS[J].Anal Chem,2011,83(20):7721-7728.
[11]Pompach P,Chandler K B,Lan R,et al.Semi-automated identification ofN-Glycopeptidesby hydrophilic interaction chromatography,nano-reverse-phase LC-MS/MS,and glycan database search[J].J Proteome Res,2012,11(3):1728-1740.
[12]Selman M H,Hemayatkar M,Deelder A M,et al.Cotton HIL⁃IC SPE microtips for microscale purification and enrichment of glycans and glycopeptides[J].Anal Chem,2011,83(7):2492-2499.
[13]Malerod H,Graham R L,Sweredoski M J,et al.Comprehen⁃sive profiling of N-linked glycosylation sites in HeLa cells us⁃ing hydrazide enrichment[J].J Proteome Res,2013,12(1):248-259.
[14]Zhang Hui,Li Xiao-jun,Martin D B.Identification and quan⁃tification of N-linked glycoproteins using hydrazide chemistry,stable isotope labeling and mass spectrometry[J].Nat Biotech⁃nol,2003,21(6):660-666.
[15]Zhong Tianping,Ai Pengfei,Zhou Jian,et al.Structures and propertiesofPAMAM dendrimer:a multi-scalesimulation study[J].Fluid Phase Equilibria,2011,302:43-47.
[16]Medina S H,El-Sayed M E H.Dendrimers as carriers for de⁃livery ofchemotherapeutic agents[J].Chem Rev,2009,109:3141-3157.
[17]Bullen H A,Hemmer R,Haskamp A.Evaluation of biotinylat⁃ed PAMAM dendrimer toxicity in models of the blood brain barrier:a biophysical and cellular approach[J].J Biomaterials Nanobiotechnol,2011,2:485-493.
[18]Domanski D M,Klajnert B,Bryszewska M.Incorporation of fluorescent probes into PAMAM dendrimers[J].Bioelectrochem⁃istry,2004,63(1):193-197.
[19]Iliuk A B,Martin V A,Alicie B M,et al.In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on met⁃al ion-functionalized soluble nanopolymers[J].Mol Cell Pro⁃teomics,2010,9:2162-2172.
[20]Kleifeld O,Doucet A,auf dem Keller U,et al.Isotopic label⁃ing of terminal amines in complex samples identifies protein N-termini and protease cleavage products[J].Nat Biotechnol,2010,3(28):281-288.
[21]Beaudette P,Nicholas A A,Huesgen P F,et al.Development of soluble ester-linked aldehyde polymers for proteomics[J].Anal Chem,2011,83:6500-6510.
[22]Wisniewski J R,Zougman A,Nagaraj N,et al.Universal sam⁃ple preparation method for proteome analysis[J].Nat Methods,2009,6:359-362.
[23]谭惠民,罗运军.树枝形聚合物[M].北京:化学工业出版社,2002:8.
[24]Dziegielewska K M,Brown W M,Casey S J,et al.The com⁃plete cDNA and amino acid sequence of bovine fetuin.Its ho⁃mologywith alpha 2HS glycoprotein and relation to other members of the cystatin superfamily[J].J Biol Chem,1990,265(8):4354-4357.
