单基因遗传病与拷贝数变异的研究进展
2014-03-29李世音秦海涛综述审校
李世音,周 浩,秦海涛(综述),郭 洪,白 云(审校)
(第三军医大学 1学员旅十五队, 2医学遗传学教研室,重庆 400038)
拷贝数变异(copy number variation,CNV)一般是指长度>1 kb的基因组大片段拷贝数增加或者减少,主要表现为亚显微水平的缺失、重复或倒位。与基因多态性(single nucleotide polymorphism,SNP)相比,CNV数量虽然不多,但涉及的基因组序列更多,突变率更高。CNV是人类遗传多样性与个体间遗传差异的一个重要原因,除了构成遗传多态性外,部分还与疾病相关。2004年,两项具有里程碑式的研究发现这种DNA拷贝数亚微观的变异广泛存在于正常人类基因组中[1-2]。Redon等[3]和Wong等[4]相继发布了人类基因组的CNV图谱。2011年,Mills等[5]指出人类基因组中每400万个碱基对中平均有超过1000个CNV。CNV是基因组结构变异的重要组成部分。近年来,CNV在遗传病中的作用越来越受到重视,除了与多基因疾病有关外,单基因病相关CNV的遗传效应也更加显著。CNV的致病机制可能与基因的剂量效应、基因断裂、基因融合以及位置效应等有关。
1 CNV的突变机制与致病机制
1.1CNV的突变机制 CNV的形成机制主要分为DNA重组与DNA错误复制两大类。DNA重组主要包括非等位同源重组和非同源末端连接[6]。非等位同源重组是不同基因组位置的两条同源重复序列配对交换导致的,主要发生在减数分裂过程中,相同染色体上重复序列间的同源重组会导致重复、缺失或倒位,而不同染色体上重复序列间的同源重组则可能导致染色体易位。与非等位同源重组不同,非同源末端连接不需要同源DNA片段作为底物,并且可以在连接部位插入碱基。
随着对CNV结构的精细解析,一些复杂的CNV无法由DNA重组模型来解释,Lee等[7]提出并证实了“复制叉停滞与模板交换”模型的存在,该模型发生在DNA复制叉停滞时,滞后链从一个复制叉上脱落,通过微同源序列转到另一个空间位置上接近的复制叉。根据复制叉的方向和位置,该模型可以导致正向或反向的缺失或重复,产生的CNV也可大可小。
1.2CNV的致病机制 染色体结构和数目畸变会导致染色体病的发生,而亚显微水平的CNV也与人类疾病密切相关[8]。CNV参与疾病的发病机制主要与相关基因的表达剂量相关,如一些剂量敏感基因的整个基因缺失或重复会导致疾病的发生。此外,如果缺失或插入发生在基因的编码区,则会导致基因断裂或融合,使蛋白产物丢失或改变。位于基因内非编码区、基因间区甚至基因荒漠区的CNV,则可能是通过位置效应影响基因的表达量而参与疾病的发生,如启动子区、非翻译区、增强子区或者隔离子区等。
2 CNV的检测方法
2.1全基因组范围的CNV研究方法 CNV作为一种直接或间接参与疾病发生、发展的新型基因组结构变异形式,如何对其进行快速、准确的检测非常重要。目前,用来进行全基因组范围CNV研究的方法有比较基因组杂交芯片技术(array-based comparative genomichybridization,aCGH)、单核苷酸多态性分型芯片技术(SNP array)和新一代测序技术(next-generation sequencing,NGS)[9]。
aCGH技术是在一张芯片上用标记不同荧光的样品(病例样品和对照样品)进行共杂交,检测病例样本基因组相对于对照基因组的DNA拷贝数变化(CNV),常用于肿瘤或遗传性疾病全基因组的CNV检测,可直观地表现出肿瘤及遗传性疾病基因组DNA在整个染色体组的缺失或扩增。
SNP array技术也是基于芯片杂交的方法,但与aCGH不同的是SNP芯片是单通道的,即一个点阵只杂交一个样本。此外,SNP芯片中的探针是专门针对单核苷酸差异设计的,因此每条探针都能得到较好的信噪比。SNP等位基因特异的探针设计提高了CNV检测灵敏度,可以鉴定中性拷贝数的杂合缺失、单亲二体及嵌合现象。早期的SNP芯片CNV探针密度非常低,但近来广泛应用的芯片(Affymetrix 6.0、cytoScan HD和Illumina 1M)中的探针更优化,针对CNV的探针也更多。因此,SNP芯片可以作为aCGH芯片的一个重要补充手段,也可以用来大规模发掘人类基因组中的CNV。
NGS技术是DNA测序技术的一次革命,它一次可以同时对几十万至几百万条DNA进行测序,故亦称为高通量测序技术。借助NGS数据也可以对CNV进行发掘、鉴定和精确定位,但需要大量的生物信息学分析,如末端配对作图、分解比对作图、序列组装及基于测序深度检测的分析方法[10-11]。虽然基于NGS方法的成本和可靠性等问题还未解决,但是该策略是今后高通量CNV分析的重要发展方向。
2.2候选CNV研究方法 全基因组CNV分析技术成本较高,无法检测到平衡结构变异,无法确定变异的具体位置,如果只对基因组中个别CNV进行研究,那么单分子候选CNV检测就显得非常合适,如荧光原位杂交、Southern blotting杂交、荧光实时定量聚合酶链反应、多重连接探针扩增以及多重基因拷贝数检测技术等。
不管是全基因组还是候选的CNV研究技术,都有优势和局限性,仅使用一种方法还不能检测出某一个体基因组所包含的全部CNV。目前为止,还未发现所有的人类基因组CNV,除了检测技术需要进一步发展外,已有的人类基因组参考序列还需要完善。
3 CNV导致的单基因遗传病
包括CNV在内的大范围的染色体结构变异一直被认为与很多人类疾病有关,CNV主要通过基因剂量效应、基因断裂、基因融合以及位置效应等方式参与疾病发生[12]。现总结了目前已知的与CNV相关的单基因遗传病(表1),以下主要依据CNV的作用区域对几种有代表性的单基因病进行介绍。
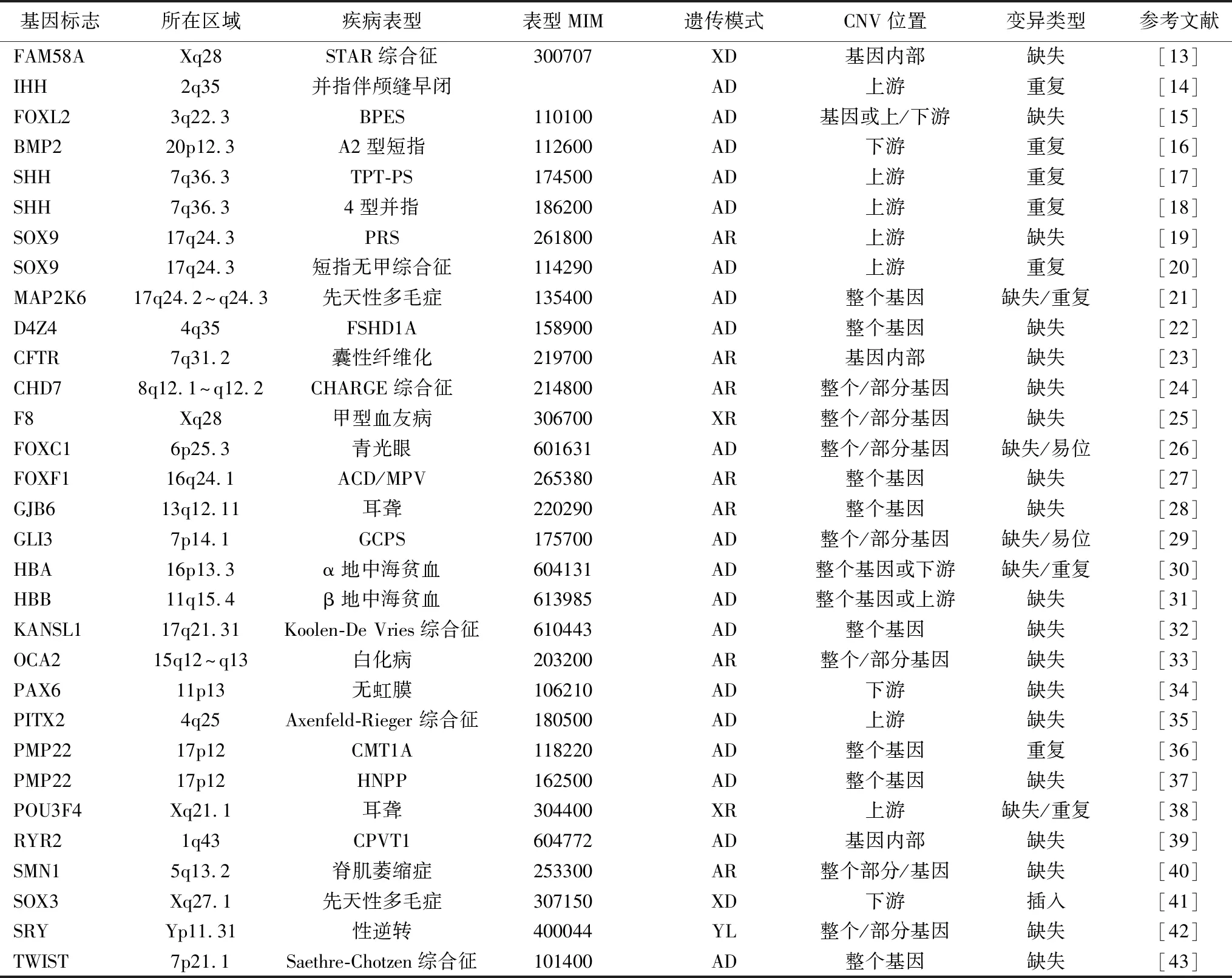
表1 CNV相关单基因遗传病
3.1CNV位于外显子编码区 并趾-内眦距增宽-肛门与生殖器异常-肾脏异常STAR综合征(toe syndactyly,telecanthus,anogenital and renal malformations,STAR综合征)是一类X染色体连锁显性遗传病,主要表现为趾尖并指、内眦距过宽、肛门生殖器及肾脏畸形。Unger等[13]通过aCGH、荧光实时定量聚合酶链反应以及测序发现,STAR综合征的主要致病原因是Xq28上FAM58A基因内的大片段缺失。FAM58A基因包含5个外显子共642 bp的编码区域,其可能参与细胞周期的调控。该基因的大片段缺失导致异常蛋白表达产物的出现,使细胞增殖受阻,从而出现相应的临床表现。
3.2CNV位于上游调控元件
3.2.1并指伴颅缝早闭 IHH基因是hedgehog家族的分泌信号分子,调控软骨细胞的分化、皮质骨的形成以及关节发育。Klopocki等[14]在颅缝早闭伴并指家系中检测到2q35区域IHH基因上游存在不同大小的重复,基因实验发现,IHH基因上游有一个高度保守的非编码元件,该元件的拷贝数增加导致了并指和颅缝早闭的发生,该元件位于邻近基因非同源末端连接1的大段内含子上,这与SHH基因的情形相似,SHH基因的增强子极化活性区调控序列(ZRS)也位于一个侧翼基因LMBR1的内含子上,说明这类基因可能有相同的进化历史,远距离调控元件在进化与发育过程中有重要作用。
3.2.2眼裂狭小综合征 眼裂狭小综合征主要表现为特征性的后仰性头部倾斜、眼裂狭小、鼻梁扁平、子宫卵巢萎缩以及眉弓隆起。D′haene等[15]在57例FOXL2基因编码区无突变的眼裂狭小综合征患者中发现,FOXL2基因上游283 kb处出现了一段长度为7.4 kb的缺失,并证实该缺失是由H-DNA介导的双链断裂所引起的。荧光素酶报告基因实验证实,该缺失区域对FOXL2基因的表达存在调控作用,并通过染色体构象捕获实验(chromosome conformation capture assay,3C实验)发现该区域与FOXL2基因启动子之间存在相互作用[15]。
3.2.3A2型短指 A2型短指临床表现为第二指和第五指中指骨畸形,主要由BMPR1B或GDF5的基因突变引起。Dathe等[16]通过连锁分析在一个A2型短指家系中定位了一个新的染色体位置20p12.3,并在BMP2基因下游110 kb处发现了一个5.5 kb高度保守的重复区域。进一步通过转基因小鼠模型发现,该区域可促进肢端X-Gal报告基因的表达。
3.3CNV位于远距离增强子
3.3.1三节拇指-并指多指综合征和4型并指 2型轴前多指的基因突变定位于7q36区域的ZRS,Lettice等[44]发现ZRS序列的点突变是2型轴前多指的致病原因。三节拇指-并指多指综合征(triphalangeal thumb-polysyndactyly syndrome,TPT-PS)和4型并指(syndactyly type 4,SD4)也定位于7q36区域。2008年,Klopocki等[17]在一个TPT-PS大家系中通过aCGH发现患者的极化活性区调控序列(ZPA regulatory sequence,ZRS)存在拷贝数重复。Sun等[18]在3个TPT-PS家系、2个伴SD4的TPT-PS家系以及1个SD4家系中同样发现患者ZRS区均存在拷贝数重复。
3.3.2腭裂与短指无甲综合征 Pierre Robin综合征是腭裂的一种,Benko等[19]发现17q24区域SOX9基因附近的微缺失与Pierre Robin综合征有关,并在体外和体内证实该缺失区域存在增强子活性,在小鼠模型中还发现该增强子调控SOX9的表达,因此推测Pierre Robin综合征的致病原因可能与该增强子的缺失引起SOX9基因的表达失控有关。随后Kurth等[20]通过aCGH在4个短指无甲综合征家系中同样发现,患者17q24.3区域均存在微重复,4个家系的最小重叠区大约为1.2 Mb,包括KCNJ2基因与SOX9基因之间的基因荒漠区,认为该区域中SOX9增强子元件的重复导致的SOX9基因时空表达紊乱可能是该病的致病原因。
3.4CNV作用于基因区域未知的单基因疾病
3.4.1先天性全身多毛症 先天性全身多毛症是一种以全身终毛过度生长、增长为表现的罕见单基因病。研究者首先在3个大家系中进行全基因组连锁分析,将致病基因定位到17q24.2~q24.3,通过基因芯片发现在该区域所有患者都存在微缺失,并在1例散发患者中,在该区域又发现了一个反向微重复,因此认为先天性全身多毛症是一种基因组疾病[21]。3个家系及散发患者CNV的最小重叠区包括4个基因,ABCA5、ABCA6、ABCA10和MAP2K6,中国汉族正常人群中存在包含这3个ABCA基因的203 kb大小的拷贝数缺失多态性,因此推测MAP2K6基因可能是先天性全身多毛症的候选致病基因,但17q24.2~q24.3区域CNV具体的生物学意义与机制仍需要进一步研究[21]。
3.4.2面肩肱型肌营养不良1型 面肩肱型肌营养不良1型是一种比较常见的遗传性肌营养不良,仅次于杜氏营养不良,大部分患者的病情进展缓慢,其特征性表现出现在20岁左右,表现为两侧面部和肩胛肌肉的显著不对称。1993年,Weiffenbach等[45]通过连锁分析将该病的致病基因定位到4q35区域,该区域位于近端粒的位置,是一个基因荒漠区,主要由3.3 kb大小的多态性重复序列D4Z4构成,正常人D4Z4的重复次数介于11~150,如果重复次数减少到10个以下,则会导致面肩肱型肌营养不良1型的发生[22]。然而时至今日,其致病基因及变异仍未发现。
4 结 语
人类基因组变异主要包含SNP、插入与缺失以及CNV等,其中CNV是人群个体间遗传差异的主要来源。CNV除了导致罕见的单基因疾病外,也与人类复杂疾病相关。目前,复杂疾病遗传学研究的瓶颈在于全基因组关联研究发现的阳性SNP位点的生物学功能难以注释,而且这些SNP位点构成的遗传效应仍然非常微小。由于CNV可能直接参与疾病的发生,其遗传效应更大,因此CNV的研究可能有助于对复杂疾病遗传机制的揭示。此外,单基因遗传病家系中致病性CNV的研究可能也有助于对复杂疾病遗传机制的理解,有助于更好地认识基因组变异与疾病间的关系。随着CNV研究方法的进展,CNV的密度和精细程度会提升,对CNV相关疾病的深入研究将对CNV的发生、效应及其对选择与进化的作用产生新的认识。
[1] Iafrate AJ,Feuk L,Rivera MN,etal.Detection of large-scale variation in the human genome[J].Nat Genet,2004,36(9):949-951.
[2] Sebat J,Lakshmi BL,Troge J,etal.Large-scale copy number polymorphism in the human genome[J].Science,2004,305(5683):525-528.
[3] Redon R,IshikawAS,Fitch KR,etal.Global variation in copy number in the human genome[J].Nature,2006,444(7118):444-454.
[4] Wong KK,deLeeuw RJ,Dosanjh NS,etal.A comprehensive analysis of common copy-number variations in the human genome[J].Am J Hum Genet,2007,80(1):91-104.
[5] Mills RE,Walter K,Stewart C,etal.Mapping copy number variation by population-scale genome sequencing[J].Nature,2011,470(7332):59-65.
[6] Liu P,Carvalho CM,Hastings PJ,etal.Mechanisms for recurrent and complex human genomic rearrangements[J].Curr Opin Genet Dev,2012,22(3):211-220.
[7] Lee JA,Carvalho CM,Lupski JR.A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders[J].Cell,2007,131(7):1235-1247.
[8] Almal SH,Padh H.Implications of gene copy-number variation in health and diseases[J].J Hum Genet,2012,57(1):6-13.
[9] Alkan C,Coe BP,Eichler EE.Genome structural variation discovery and genotyping[J].Nat Rev Genet,2011,12(5):363-376.
[10] Korbel JO,Urban AE,Affourtit JP,etal.Paired-end mapping reveals extensive structural variation in the human genome[J].Science,2007,318(5849):420-426.
[11] Sudmant PH,Kitzman JO,Antonacci F,etal.Diversity of human copy number variation and multicopy genes[J].Science,2010,330(6004):641-646.
[12] Malhotra D,Sebat J.CNVs:harbingers of a rare variant revolution in psychiatric genetics[J].Cell,2012,148(6):1223-1241.
[13] Unger S,Böhm D,Kaiser FJ,etal.Mutations in the cyclin family member FAM58A cause an X-linked dominant disorder characterized by syndactyly,telecanthus and anogenital and renal malformations[J].Nat Genet,2008,40(3):287-289.
[14] Klopocki E,Lohan S,Brancati F,etal.Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis[J].Am J Hum Genet,2011,88(1):70-75.
[15] D′haene B,Attanasio C,Beysen D,etal.Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor:implications for mutation screening[J].PLoS Genet,2009,5(6):e1000522.
[16] Dathe K,Kjaer KW,Brehm A,etal.Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2[J].Am J Hum Genet,2009,84(4):483-492.
[17] Klopocki E,Ott CE,Benatar N,etal.A microduplication of the long range SHH limb regulator(ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome[J].J Med Genet,2008,45(6):370-375.
[18] Sun M,Ma F,Zeng X,etal.Triphalangeal thumb-polysyndactyly syndrome and syndactyly type Ⅳ are caused by genomic duplications involving the long range,limb-specific SHH enhancer[J].J Med Genet,2008,45(9):589-595.
[19] Benko S,Fantes JA,Amiel J,etal.Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence[J].Nat Genet,2009,41(3):359-364.
[20] Kurth I,Klopocki E,Stricker S,etal.Duplications of noncoding elements 5′ of SOX9 are associated with brachydactyly-anonychia[J].Nat Genet,2009,41(8):862-863.
[21] Sun M,Li N,Dong W,etal.Copy-number mutations on chromosome 17q24.2-q24.3 in congenital generalized hypertrichosis terminalis with or without gingival hyperplasia[J].Am J Hum Genet,2009,84(6):807-813.
[22] Cabianca DS,Gabellini D.The cell biology of disease:FSHD:copy number variations on the theme of muscular dystrophy[J].J Cell Biol,2010,191(6):1049-1060.
[23] Audrézet MP,Chen JM,Raguénès O,etal.Genomic rearrangements in the CFTR gene:extensive allelic heterogeneity and diverse mutational mechanisms[J].Hum Mutat,2004,23(4):343-357.
[24] Vissers LE,van Ravenswaaij CM,Admiraal R,etal.Mutations in a new member of the chromodomain gene family cause CHARGE syndrome[J].Nat Genet,2004,36(9):955-957.
[25] Green PM,Bagnall RD,Waseem NH,etal.Haemophilia A mutations in the UK:results of screening one-third of the population[J].Br J Haematol,2008,143(1):115-128.
[26] Chanda B,Asai-Coakwell M,Ye M,etal.A novel mechanistic spectrum underlies glaucoma-associated chromosome 6p25 copy number variation[J].Hum Mol Genet,2008,17(22):3446-3458.
[27] Stankiewicz P,Sen P,Bhatt SS,etal.Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations[J].Am J Hum Genet,2009,84(6):780-791.
[28] del Castillo FJ,Rodríguez-Ballesteros M,Alvarez A,etal.A novel deletion involving the connexin-30 gene,del(GJB6-d13s1854),found in trans with mutations in the GJB2 gene(connexin-26) in subjects with DFNB1 non-syndromic hearing impairment[J].J Med Genet,2005,42(7):588-594.
[29] Hurst JA,Jenkins D,Vasudevan PC,etal.Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome:five cases with intragenic mutations or complete deletions of GLI3[J].Eur J Hum Genet,2011,19(7):757-762.
[30] Liao YM,Lin SK,Liu TC,etal.Rapid identification of the copy number of α-globin genes by capillary electrophoresis analysis[J].Clin Biochem,2012,45(10/11):798-805.
[31] Babashah S,Jamali S,Mahdian R,etal.Detection of unknown deletions in beta-globin gene cluster using relative quantitative PCR methods[J].Eur J Haematol,2009,83(3):261-269.
[32] Zollino M,Orteschi D,Murdolo M,etal.Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype[J].Nat Genet,2012,44(6):636-638.
[33] Rooryck C,Morice-Picard F,Lasseaux E,etal.High resolution mapping of OCA2 intragenic rearrangements and identification of a founder effect associated with a deletion in Polish albino patients[J].Hum Genet,2011,129(2):199-208.
[34] D′Elia AV,Pellizzari L,Fabbro D,etal.A deletion 3′ to the PAX6 gene in familial aniridia cases[J].Mol Vis,2007,13:1245-1250.
[35] Flomen RH,Gorman PA,Vatcheva R,etal.Rieger syndrome locus:a new reciprocal translocation t(4;12)(q25;q15) and a deletion del(4)(q25q27) both break between markers D4S2945 and D4S193[J].J Med Genet,1997,34(3):191-195.
[36] Weterman MA,van Ruissen F,de Wissel M,etal.Copy number variation upstream of PMP22 in Charcot-Marie-Tooth disease[J].Eur J Hum Genet,2010,18(4):421-428.
[37] Li J,Ghandour K,Radovanovic D,etal.Stoichiometric alteration of PMP22 protein determines the phenotype of hereditary neuropathy with liability to pressure palsies[J].Arch Neurol,2007,64(7):974-978.
[38] de Kok YJ,van der Maarel SM,Bitner-Glindzicz M,etal.Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4[J].Science,1995,267(5198):685-688.
[39] Bhuiyan ZA,van den Berg MP,van Tintelen JP,etal.Expanding spectrum of human RYR2-related disease:new electrocardiographic,structural,and genetic features[J].Circulation,2007,116(14):1569-1576.
[40] Alías L,Bernal S,Fuentes-Prior P,etal.Mutation update of spinal muscular atrophy in Spain:molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene[J].Hum Genet,2009,125(1):29-39.
[41] Zhu H,Shang D,Sun M,etal.X-linked congenital hypertrichosis syndrome is associated with interchromosomal insertions mediated by a human-specific palindrome near SOX3[J].Am J Hum Genet,2011,88(6):819-826.
[42] Lange J,Skaletsky H,van Daalen SK,etal.Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes[J].Cell,2009,138(5):855-869.
[43] Chun K,Teebi AS,Jung JH,etal.Genetic analysis of patients with the Saethre-Chotzen phenotype[J].Am J Med Genet,2002,110(2):136-143.
[44] Lettice LA,Heaney SJ,Purdie LA,etal.A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly[J].Hum Mol Genet,2003,12(14):1725-1735.
[45] Weiffenbach B,Dubois J,Storvick D,etal.Mapping the facioscapulohumeral muscular dystrophy gene is complicated by chromsome 4q35 recombination events[J].Nat Genet,1993,4(2):165-169.
